
Transcriptome-Wide Association Study of Blood Cell Traits in African Ancestry and Hispanic/Latino Populations

 , , , , , , , , ,
, , , , , , , , ,
Abstract
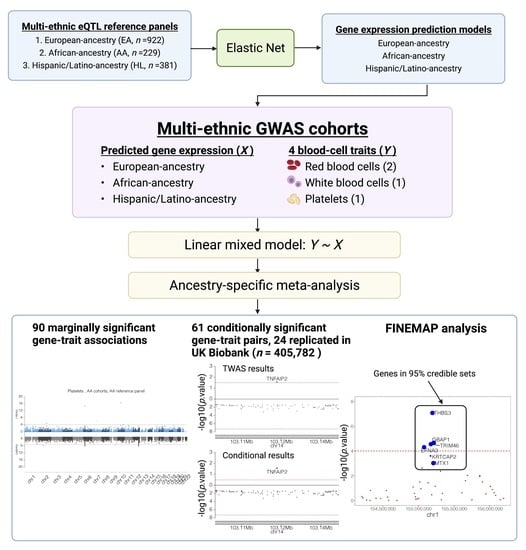
:
1. Introduction
2. Materials and Methods
2.1. Training of TWAS Prediction Models
2.2. Assessment of Trained Expression Prediction Models in GENOA
2.3. Phenotype
2.4. TWAS Association and Conditional Analysis
2.5. Replication
3. Results
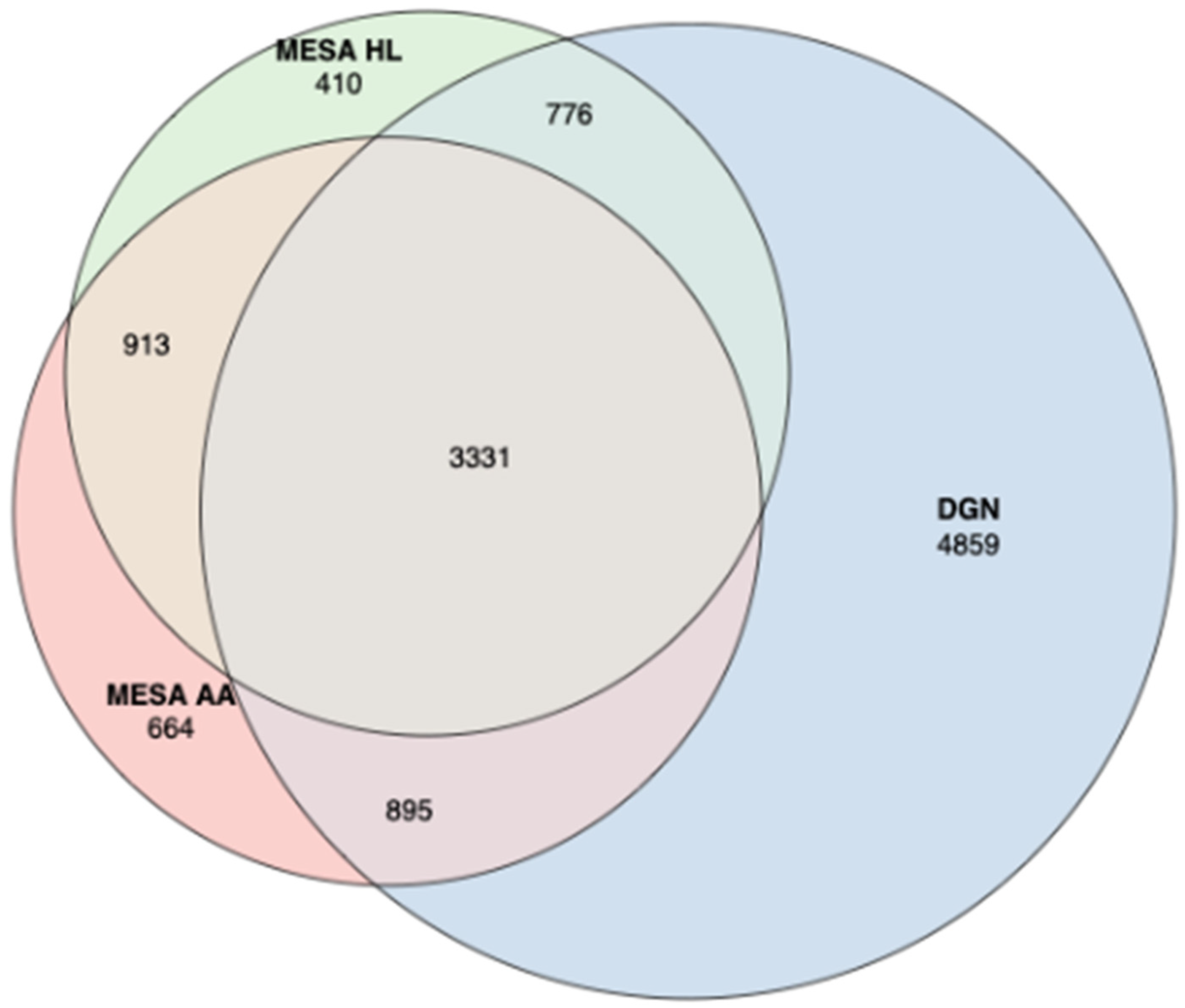
3.1. Train Gene Expression Prediction Models from Reference eQTL Datasets
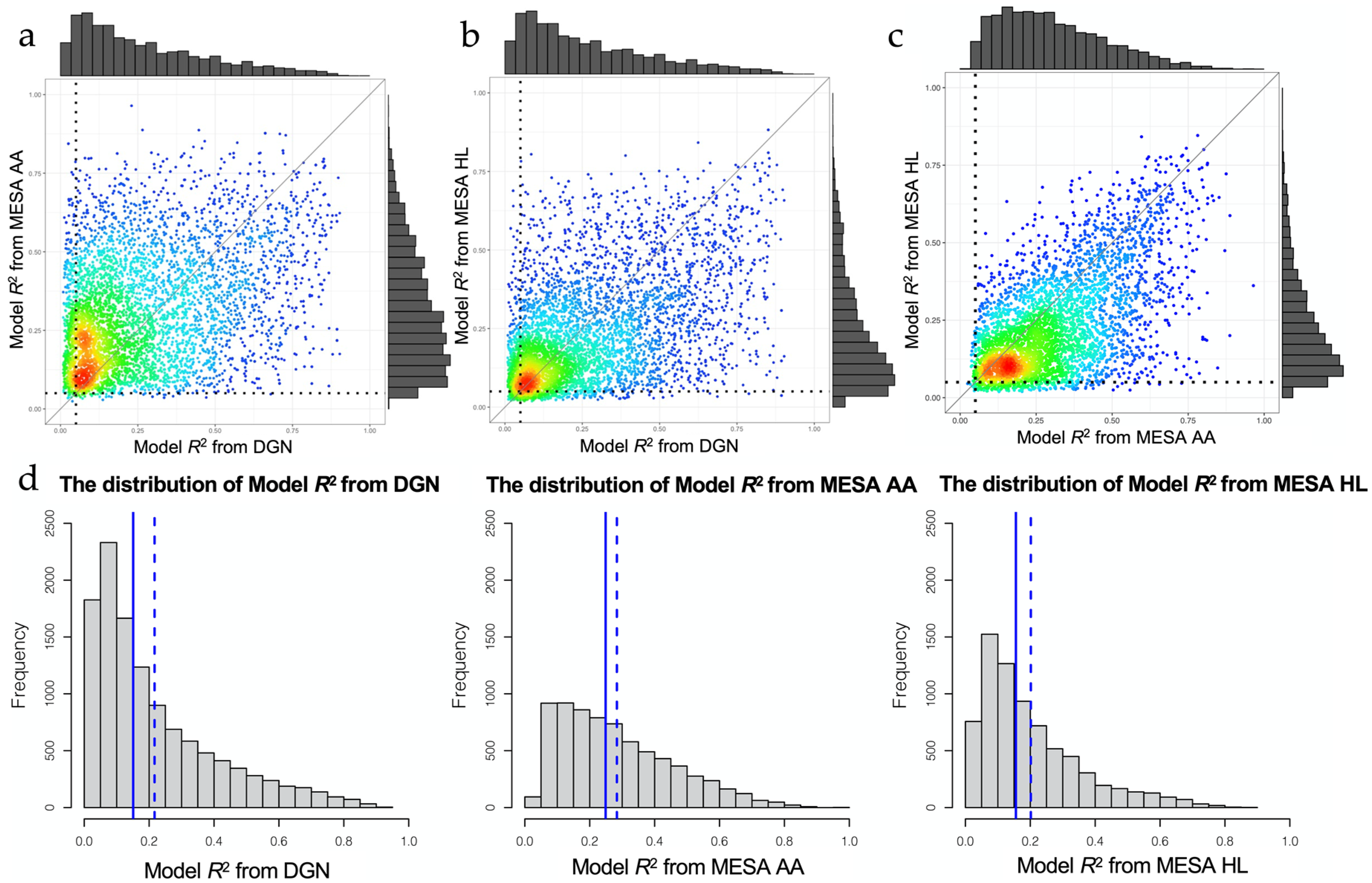
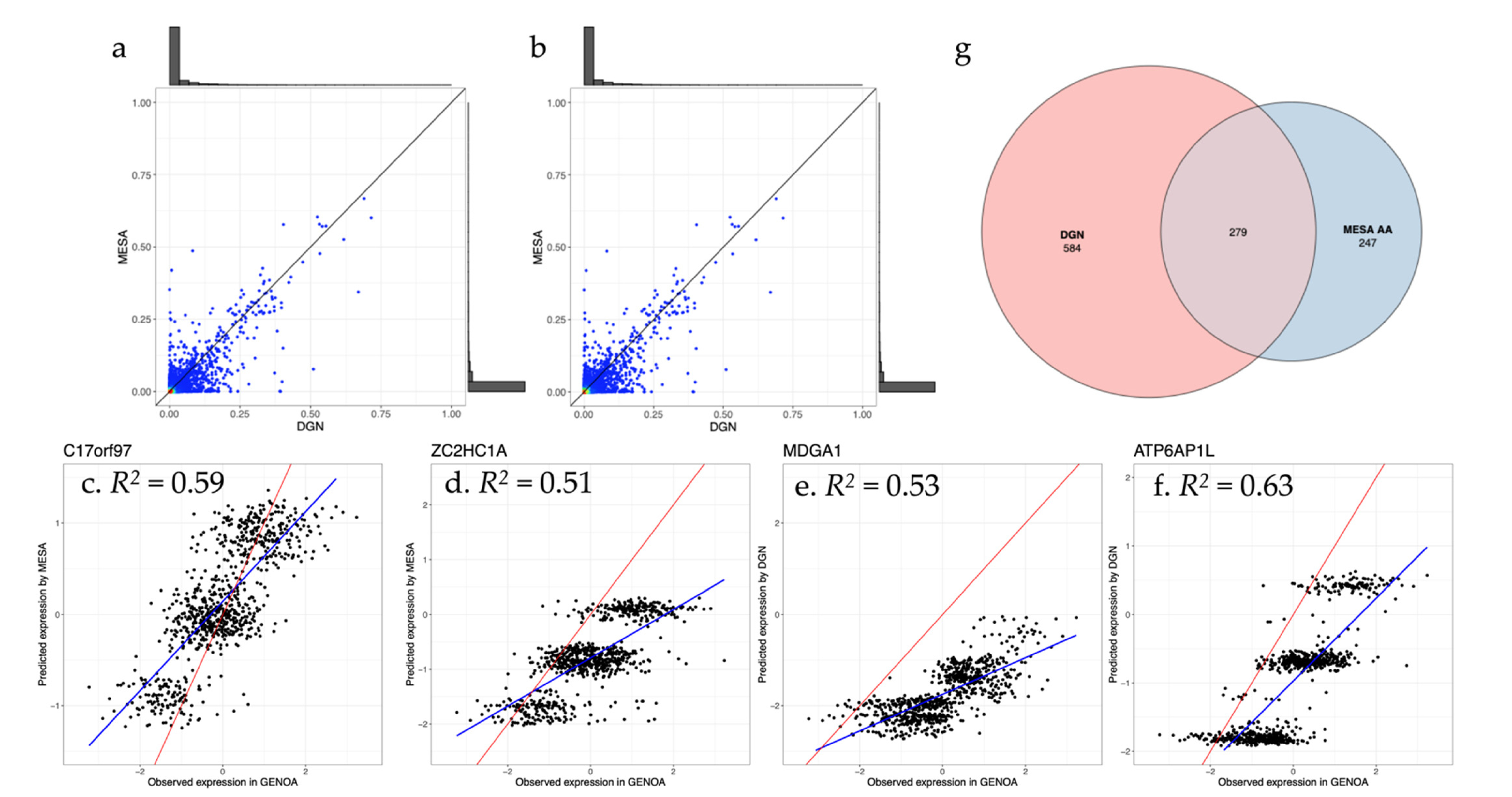
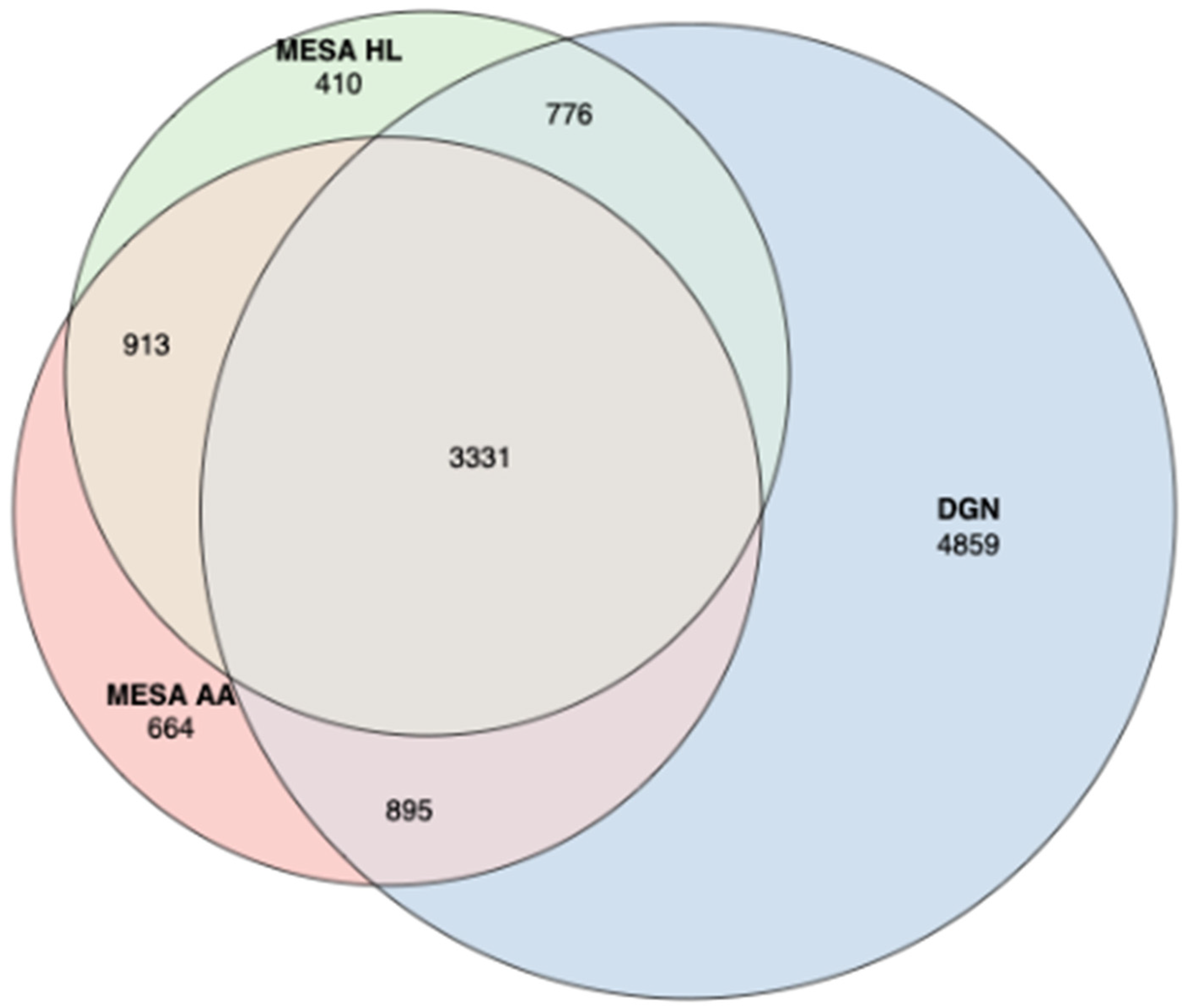
3.2. Assessment of Trained Expression Prediction Models in Independent Non-European Datasets
3.3. Association between Predicted Gene Expression and Blood independent Traits
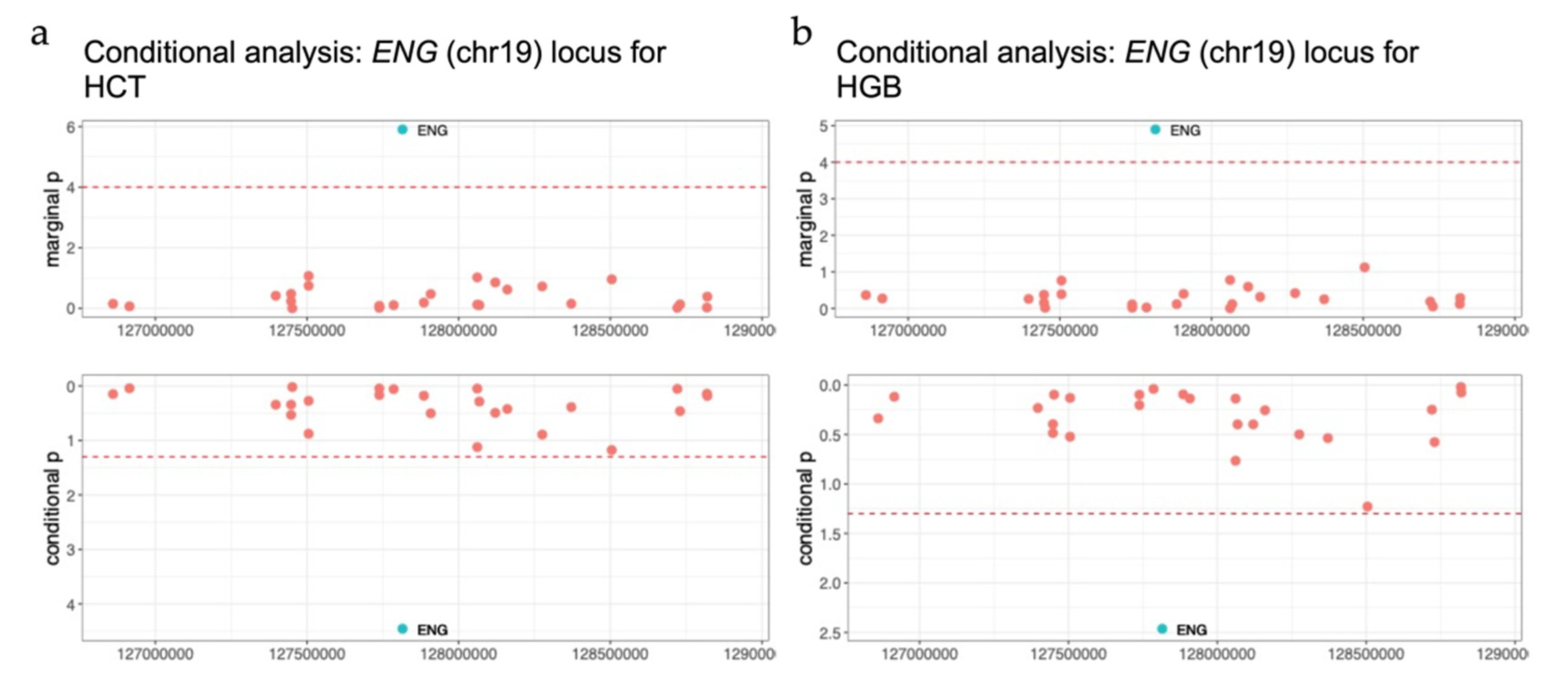
3.4. TWAS Analysis Conditional on Neighboring GWAS Variants
3.5. Replication in UK Biobank
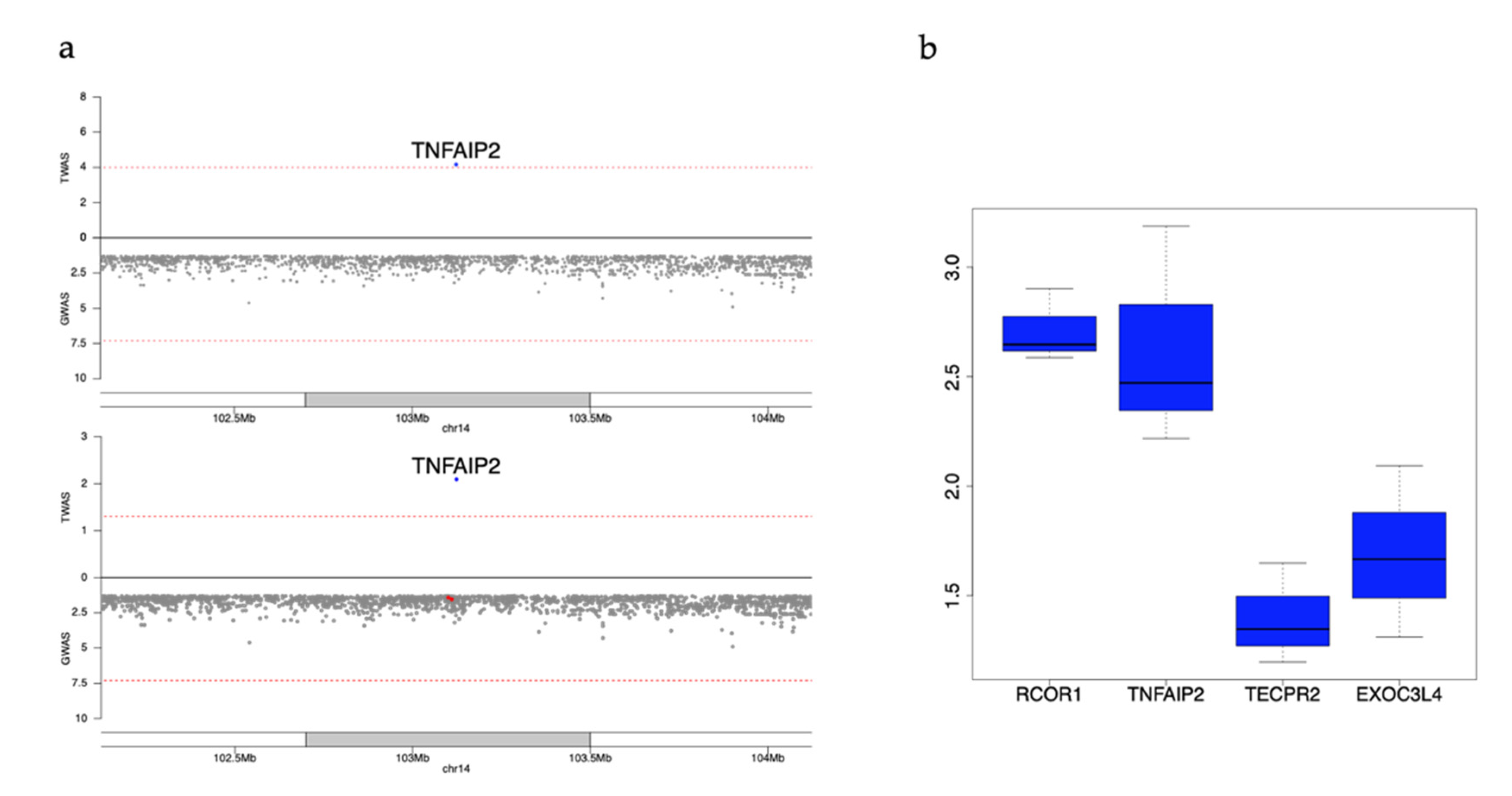
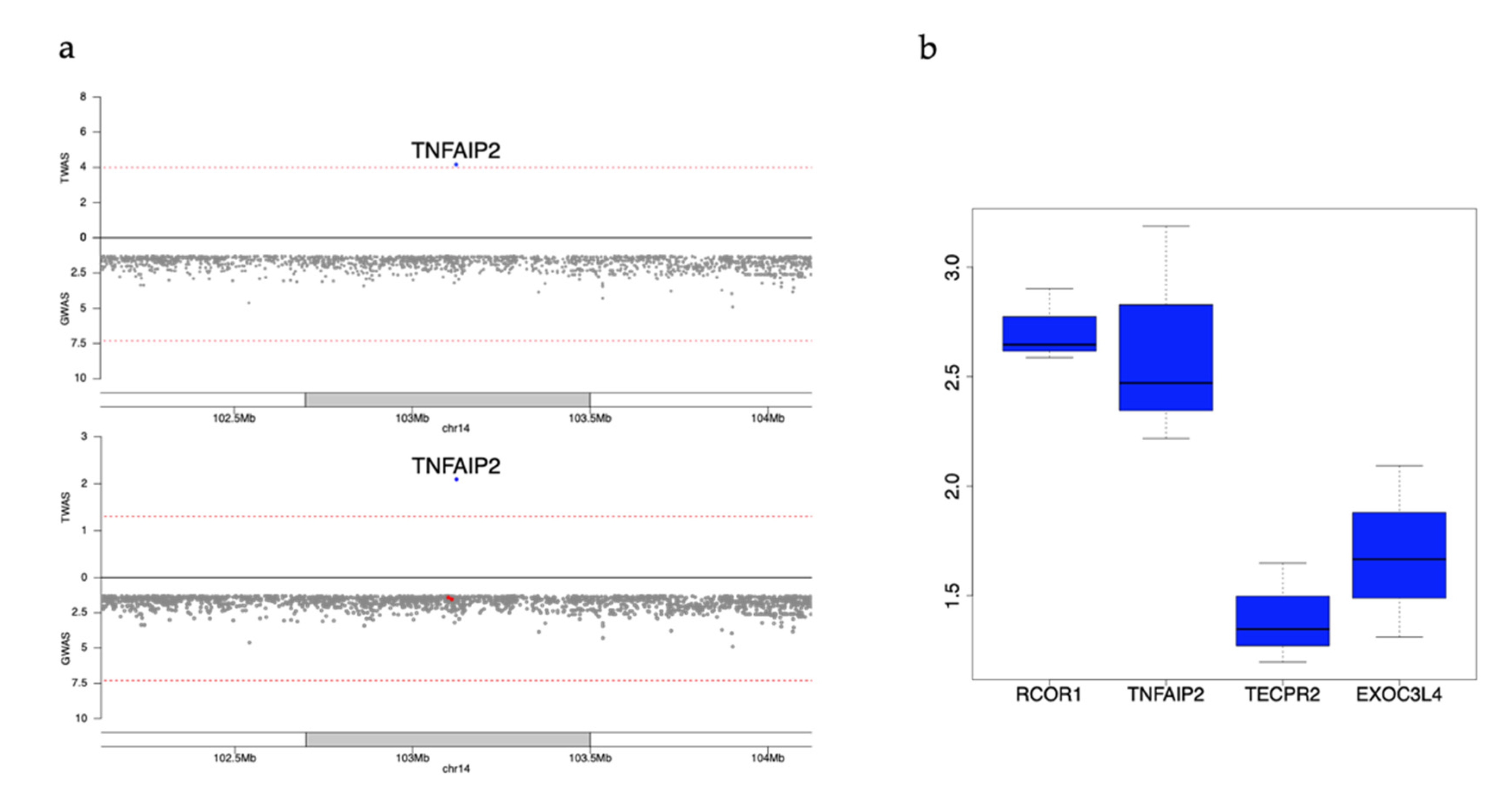
3.6. Example Replicated Genes Still Nominally Significant after Conditioning on Known GWAS Variants
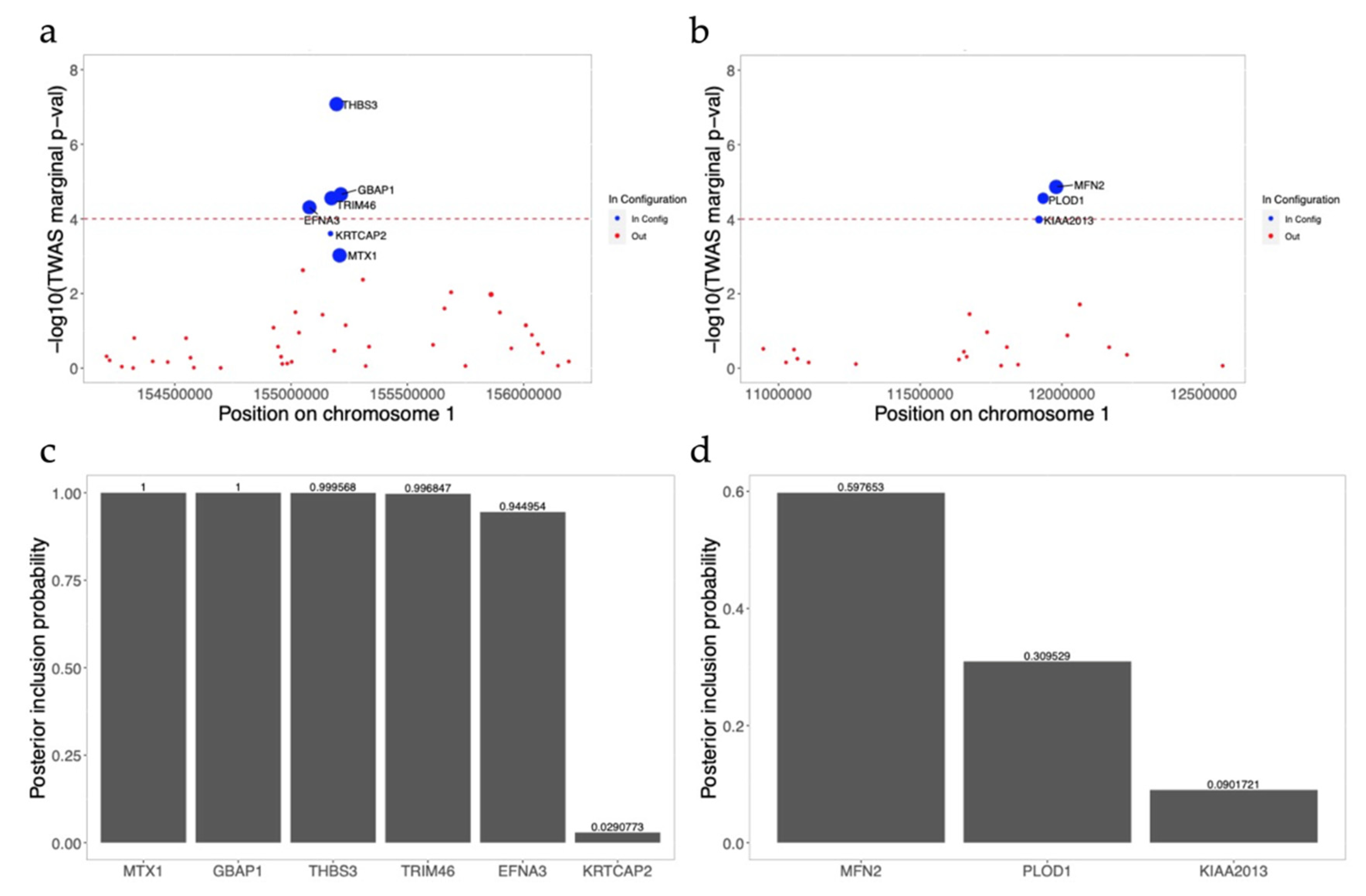
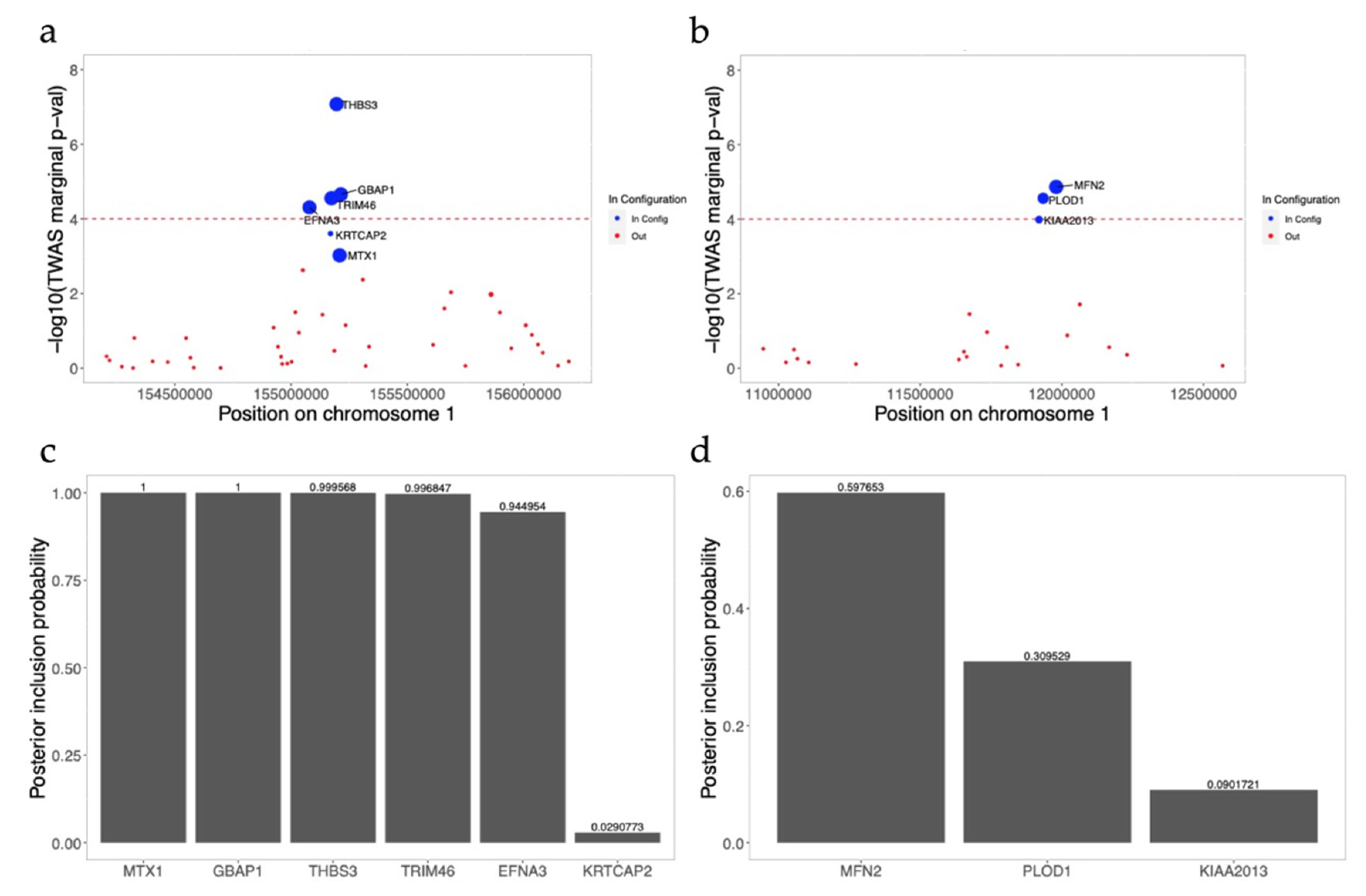
3.7. FINEMAP Analysis for Significant Gene-Trait Associations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Segal, J.B.; Moliterno, A.R. Platelet Counts Differ by Sex, Ethnicity, and Age in the United States. Ann. Epidemiology 2006, 16, 123–130. [Google Scholar] [CrossRef]
- Zakai, N.A.; McClure, L.A.; Prineas, R.; Howard, G.; McClellan, W.; Holmes, C.E.; Newsome, B.B.; Warnock, D.G.; Audhya, P.; Cushman, M. Correlates of Anemia in American Blacks and Whites: The REGARDS Renal Ancillary Study. Am. J. Epidemiology 2008, 169, 355–364. [Google Scholar] [CrossRef]
- Lim, E.-M.; Cembrowski, G.; Clarke, G. Race-specific WBC and neutrophil count reference intervals. Int. J. Lab. Hematol. 2010, 32, 590–597. [Google Scholar] [CrossRef]
- Reich, D.; Nalls, M.A.; Kao, W.H.L.; Akylbekova, E.L.; Tandon, A.; Patterson, N.; Mullikin, J.; Hsueh, W.-C.; Cheng, C.-Y.; Coresh, J.; et al. Reduced Neutrophil Count in People of African Descent Is Due To a Regulatory Variant in the Duffy Antigen Receptor for Chemokines Gene. PLoS Genet. 2009, 5, e1000360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beutler, E.; West, C. Hematologic differences between African-Americans and whites: The roles of iron deficiency and α-thalassemia on hemoglobin levels and mean corpuscular volume. Blood 2005, 106, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Network, M.G.E. Insights into malaria susceptibility using genome-wide data on 17,000 individuals from Africa, Asia and Oceania. Nat. Commun. 2019, 10, 5732. [Google Scholar] [CrossRef] [Green Version]
- Clarke, G.M.; Rockett, K.; Kivinen, K.; Hubbart, C.; Jeffreys, A.E.; Rowlands, K.; Jallow, M.; Conway, D.; Bojang, K.A.; Pinder, M.; et al. Characterisation of the opposing effects of G6PD deficiency on cerebral malaria and severe malarial anaemia. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Stilp, A.M.; McHugh, C.P.; Rao, S.; Jain, D.; Zheng, X.; Lane, J.; de Bellefon, S.M.; Raffield, L.M.; Chen, M.-H.; et al. Whole-genome sequencing association analysis of quantitative red blood cell phenotypes: The NHLBI TOPMed program. Am. J. Hum. Genet. 2021, 108, 874–893. [Google Scholar] [CrossRef]
- Astle, W.; Elding, H.; Jiang, T.; Allen, D.; Ruklisa, D.; Mann, A.; Mead, D.; Bouman, H.; Riveros-Mckay, F.; Kostadima, M.A.; et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 2016, 167, 1415–1429.e19. [Google Scholar] [CrossRef] [Green Version]
- Ganesh, S.K.; A Zakai, N.; A Van Rooij, F.J.; Soranzo, N.; Smith, A.V.; A Nalls, M.; Chen, M.-H.; Kottgen, A.; Glazer, N.; Dehghan, A.; et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat. Genet. 2009, 41, 1191–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuckovic, D.; Bao, E.L.; Akbari, P.; Lareau, C.A.; Mousas, A.; Jiang, T.; Chen, M.-H.; Raffield, L.M.; Tardaguila, M.; Huffman, J.E.; et al. The Polygenic and Monogenic Basis of Blood Traits and Diseases. Cell 2020, 182, 1214–1231.e11. [Google Scholar] [CrossRef] [PubMed]
- Popejoy, A.B.; Fullerton, S.M. Genomics is failing on diversity. Nat. Cell Biol. 2016, 538, 161–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peprah, E.; Wiley, K.; Sampson, U.; Narula, J. A New Age for African-Driven Genomics Research: Human Heredity and Health in Africa (H3Africa). Glob. Hear. 2017, 12, 67–68. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, M.; Sinnott-Armstrong, N.; Mancuso, N.; Barbeira, A.N.; Knowles, D.A.; Golan, D.; Ermel, R.; Ruusalepp, A.; Quertermous, T.; Hao, K.; et al. Opportunities and challenges for transcriptome-wide association studies. Nat. Genet. 2019, 51, 592–599. [Google Scholar] [CrossRef]
- Gamazon, E.R.; GTEx Consortium; Wheeler, H.; Shah, K.P.; Mozaffari, S.; Aquino-Michaels, K.; Carroll, R.J.; Eyler, A.E.; Denny, J.C.; Nicolae, D.; et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015, 47, 1091–1098. [Google Scholar] [CrossRef] [Green Version]
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.J.H.; Jansen, R.; De Geus, E.J.C.; I Boomsma, D.; A Wright, F.; et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016, 48, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Barbeira, A.N.; GTEx Consortium; Dickinson, S.P.; Bonazzola, R.; Zheng, J.; Wheeler, H.; Torres, J.M.; Torstenson, E.S.; Shah, K.P.; Garcia, T.; et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 2018, 9, 1–20. [Google Scholar] [CrossRef]
- Wu, L.; Shi, W.; Long, J.; Guo, X.; Michailidou, K.; Beesley, J.; Bolla, M.K.; Shu, X.-O.; Lu, Y.; Cai, Q.; et al. A transcriptome-wide association study of 229,000 women identifies new candidate susceptibility genes for breast cancer. Nat. Genet. 2018, 50, 968–978. [Google Scholar] [CrossRef]
- Mancuso, N.; The PRACTICAL Consortium; Gayther, S.; Gusev, A.; Zheng, W.; Penney, K.L.; Kote-Jarai, Z.; Eeles, R.; Freedman, M.; Haiman, C.; et al. Large-scale transcriptome-wide association study identifies new prostate cancer risk regions. Nat. Commun. 2018, 9, 4079. [Google Scholar] [CrossRef] [Green Version]
- Andaleon, A.; Mogil, L.S.; Wheeler, H.E. Genetically regulated gene expression underlies lipid traits in Hispanic cohorts. PLoS ONE 2019, 14, e0220827. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; García-Closas, M.; Olshan, A.F.; Perou, C.M.; Troester, M.A.; Love, M.I. A framework for transcriptome-wide association studies in breast cancer in diverse study populations. Genome Biol. 2020, 21, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.; García-Closas, M.; Olshan, A.F.; Perou, C.M.; Troester, M.A.; Love, M.I.; Bhattacharya, A. Gene-level germline contributions to clinical risk of recurrence scores in Black and White breast cancer patients. medRxiv 2021. [Google Scholar] [CrossRef]
- Fiorica, P.N.; Schubert, R.; Morris, J.D.; Sami, M.A.; Wheeler, H.E. Multi-ethnic transcriptome-wide association study of prostate cancer. PLoS ONE 2020, 15, e0236209. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, E.; Gregga, I.; Wheeler, H.E. Population-Matched Transcriptome Prediction Increases TWAS Discovery and Replication Rate. iScience 2020, 23, 101850. [Google Scholar] [CrossRef]
- Ki Sung GTEx Consortium. Genetic effects on gene expression across human tissues. Nat. Cell Biol. 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Battle, A.; Mostafavi, S.; Zhu, X.; Potash, J.B.; Weissman, M.; McCormick, C.; Haudenschild, C.D.; Beckman, K.B.; Shi, J.; Mei, R.; et al. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 2014, 24, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Forer, L.; Schöenherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [Green Version]
- Maples, B.K.; Gravel, S.; Kenny, E.; Bustamante, C.D. RFMix: A Discriminative Modeling Approach for Rapid and Robust Local-Ancestry Inference. Am. J. Hum. Genet. 2013, 93, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Mostafavi, S.; Battle, A.; Zhu, X.; Urban, A.E.; Levinson, D.; Montgomery, S.B.; Koller, D. Normalizing RNA-Sequencing Data by Modeling Hidden Covariates with Prior Knowledge. PLoS ONE 2013, 8, e68141. [Google Scholar] [CrossRef]
- Mogil, L.S.; Andaleon, A.; Badalamenti, A.; Dickinson, S.P.; Guo, X.; Rotter, J.I.; Johnson, W.C.; Im, H.K.; Liu, Y.; Wheeler, H.E. Genetic architecture of gene expression traits across diverse populations. PLoS Genet. 2018, 14, e1007586. [Google Scholar] [CrossRef]
- Liu, Y.; Reynolds, L.M.; Ding, J.; Hou, L.; Lohman, K.; Young, T.; Cui, W.; Huang, Z.; Grenier, C.; Wan, M.; et al. Blood monocyte transcriptome and epigenome analyses reveal loci associated with human atherosclerosis. Nat. Commun. 2017, 8, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ding, J.; Reynolds, L.; Lohman, K.; Register, T.C.; De La Fuente, A.; Howard, T.D.; Hawkins, G.A.; Cui, W.; Morris, J.; et al. Methylomics of gene expression in human monocytes. Hum. Mol. Genet. 2013, 22, 5065–5074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegle, O.; Parts, L.; Piipari, M.; Winn, J.; Durbin, R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 2012, 7, 500–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The 1000 Genomes Project Consortium A global reference for human genetic variation. Nature 2015, 526, 68–74. [CrossRef] [Green Version]
- Shang, L.; Smith, J.A.; Zhao, W.; Kho, M.; Turner, S.T.; Mosley, T.H.; Kardia, S.L.; Zhou, X. Genetic Architecture of Gene Expression in European and African Americans: An eQTL Mapping Study in GENOA. Am. J. Hum. Genet. 2020, 106, 496–512. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Li, Y.; Abecasis, G.R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010, 26, 2190–2191. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, S.; Li, Z.; Morrison, A.C.; Boerwinkle, E.; Lin, X. ACAT: A Fast and Powerful p Value Combination Method for Rare-Variant Analysis in Sequencing Studies. Am. J. Hum. Genet. 2019, 104, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-H.; Raffield, L.M.; Mousas, A.; Sakaue, S.; Huffman, J.E.; Moscati, A.; Trivedi, B.; Jiang, T.; Akbari, P.; Vuckovic, D.; et al. Trans-ethnic and Ancestry-Specific Blood-Cell Genetics in 746,667 Individuals from 5 Global Populations. Cell 2020, 182, 1198–1213.e14. [Google Scholar] [CrossRef]
- Benner, C.; Spencer, C.; Havulinna, A.S.; Salomaa, V.; Ripatti, S.; Pirinen, M. FINEMAP: Efficient variable selection using summary data from genome-wide association studies. Bioinformatics 2016, 32, 1493–1501. [Google Scholar] [CrossRef] [Green Version]
- Mbatchou, J.; Barnard, L.; Backman, J.; Marcketta, A.; Kosmicki, J.A.; Ziyatdinov, A.; Benner, C.; O’Dushlaine, C.; Barber, M.; Boutkov, B.; et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 2021, 1–7. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Ulirsch, J.C.; Lareau, C.A.; Bao, E.L.; Ludwig, L.S.; Guo, M.H.; Benner, C.; Satpathy, A.T.; Kartha, V.; Salem, R.M.; Hirschhorn, J.N.; et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat. Genet. 2019, 51, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Javierre, B.M.; Burren, O.S.; Wilder, S.; Kreuzhuber, R.; Hill, S.M.; Sewitz, S.; Cairns, J.; Wingett, S.; Varnai, C.; Thiecke, M.J.; et al. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 2016, 167, 1369–1384.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kichaev, G.; Bhatia, G.; Loh, P.-R.; Gazal, S.; Burch, K.; Freund, M.K.; Schoech, A.; Pasaniuc, B.; Price, A.L. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am. J. Hum. Genet. 2019, 104, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieger, C.; Radhakrishnan, A.; Cvejic, A.; Tang, W.; Porcu, E.; Pistis, G.; Serbanovic-Canic, J.; Elling, U.; Goodall, A.; Labrune, Y.; et al. New gene functions in megakaryopoiesis and platelet formation. Nat. Cell Biol. 2011, 480, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Wilson, J.G.; Patterson, N.J.; Tandon, A.; Zmuda, J.M.; Huntsman, S.; Garcia, M.; Hu, D.; Li, R.; Beamer, B.A.; et al. Admixture Mapping of White Cell Count: Genetic Locus Responsible for Lower White Blood Cell Count in the Health ABC and Jackson Heart Studies. Am. J. Hum. Genet. 2008, 82, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Genovese, G.; Friedman, D.J.; Ross, M.D.; Lecordier, L.; Uzureau, P.; Freedman, B.I.; Bowden, D.W.; Langefeld, C.D.; Oleksyk, T.K.; Knob, A.L.U.; et al. Association of Trypanolytic ApoL1 Variants with Kidney Disease in African Americans. Science 2010, 329, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedman, B.I.; Moxey-Mims, M.M.; Alexander, A.A.; Astor, B.C.; Birdwell, K.A.; Bowden, D.W.; Bowen, G.; Bromberg, J.; Craven, T.E.; Dadhania, D.M.; et al. APOL1 Long-term Kidney Transplantation Outcomes Network (APOLLO): Design and Rationale. Kidney Int. Rep. 2020, 5, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Traylor, M.; Lewis, C.M. Genetic discovery in multi-ethnic populations. Eur. J. Hum. Genet. 2016, 24, 1097–1098. [Google Scholar] [CrossRef] [Green Version]
- Wojcik, G.L.; Graff, M.; Nishimura, K.K.; Tao, R.; Haessler, J.; Gignoux, C.R.; Highland, H.M.; Patel, Y.M.; Sorokin, E.P.; Avery, C.L.; et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 2019, 570, 514–518. [Google Scholar] [CrossRef]
- Hurst, L.D. Genetics and the understanding of selection. Nat. Rev. Genet. 2009, 10, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.J.; Williams, J.S.; Egede, L.E. Influence of Race, Ethnicity and Social Determinants of Health on Diabetes Outcomes. Am. J. Med. Sci. 2016, 351, 366–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogungbe, O.; Turkson-Ocran, R.-A.; Nkimbeng, M.; Cudjoe, J.; Miller, H.N.; Baptiste, D.; Himmelfarb, C.D.; Davidson, P.; Cooper, L.A.; Commodore-Mensah, Y. Social determinants of hypertension and diabetes among African immigrants: The African immigrants health study. Ethn. Heal. 2021, 1–13. [Google Scholar] [CrossRef]
- Hodonsky, C.J.; Baldassari, A.R.; Bien, S.A.; Raffield, L.M.; Highland, H.M.; Sitlani, C.M.; Wojcik, G.L.; Tao, R.; Graff, M.; Tang, W.; et al. Ancestry-specific associations identified in genome-wide combined-phenotype study of red blood cell traits emphasize benefits of diversity in genomics. BMC Genom. 2020, 21, 228. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, M.H.; Qian, H.; Hou, Z.; Rosen, J.D.; Tapia, A.L.; Shan, Y.; Jain, D.; Argos, M.; Arnett, D.K.; Avery, C.; et al. Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 2019, 15, e1008500. [Google Scholar] [CrossRef] [Green Version]
- Keys, K.L.; Mak, A.C.Y.; White, M.J.; Eckalbar, W.L.; Dahl, A.W.; Mefford, J.; Mikhaylova, A.; Contreras, M.G.; Elhawary, J.R.; Eng, C.; et al. On the cross-population generalizability of gene expression prediction models. PLoS Genet. 2020, 16, e1008927. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Kasela, S.; et al. Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv 2018, 447367. [Google Scholar] [CrossRef] [Green Version]
- Ramsingh, G.; Koboldt, D.C.; Trissal, M.; Chiappinelli, K.B.; Wylie, T.; Koul, S.; Chang, L.-W.; Nagarajan, R.; Fehniger, T.A.; Goodfellow, P.; et al. Complete characterization of the microRNAome in a patient with acute myeloid leukemia. Blood 2010, 116, 5316–5326. [Google Scholar] [CrossRef] [Green Version]
- Rusiniak, M.E.; Yu, M.; Ross, D.T.; Tolhurst, E.C.; Slack, J.L. Identification of B94 (TNFAIP2) as a potential retinoic acid target gene in acute promyelocytic leukemia. Cancer Res. 2000, 60, 1824–1829. [Google Scholar]
- Zhao, D.; Deng, S.-C.; Ma, Y.; Hao, Y.-H.; Jia, Z.-H. miR-221 alleviates the inflammatory response and cell apoptosis of neuronal cell through targeting TNFAIP2 in spinal cord ischemia–reperfusion. NeuroReport 2018, 29, 655–660. [Google Scholar] [CrossRef]
- Sarma, V.; Wolf, F.W.; Marks, R.M.; Shows, T.B.; Dixit, V.M. Cloning of a novel tumor necrosis factor-alpha-inducible primary response gene that is differentially expressed in development and capillary tube-like formation in vitro. J. Immunol. 1992, 148, 3302–3312. [Google Scholar] [PubMed]
- Qu, X.; Li, Q.; Zhang, X.; Wang, Z.; Wang, S.; Zhou, Z. Amentoflavone protects the hematopoietic system of mice against γ-irradiation. Arch. Pharmacal Res. 2019, 42, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.; Sarma, V.; Seldin, M.; Drake, S.; Suchard, S.; Shao, H.; O’Shea, K.; Dixit, V. B94, a primary response gene inducible by tumor necrosis factor-alpha, is expressed in developing hematopoietic tissues and the sperm acrosome. J. Biol. Chem. 1994, 269, 3633–3640. [Google Scholar] [CrossRef]
- Cheng, Z.; Wang, H.Z.; Li, X.; Wu, Z.; Han, Y.; Li, Y.; Chen, G.; Xie, X.; Huang, Y.; Du, Z.; et al. MicroRNA-184 inhibits cell proliferation and invasion, and specifically targets TNFAIP2 in Glioma. J. Exp. Clin. Cancer Res. 2015, 34, 27. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wei, S.; Ma, H.; Zhao, M.; Myers, J.N.; Weber, R.S.; Sturgis, E.M.; Wei, Q. A functional variant at the miR-184 binding site in TNFAIP2 and risk of squamous cell carcinoma of the head and neck. Carcinogenesis 2011, 32, 1668–1674. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Chr | Start_hg38 | End_hg38 | Phenotype | Meta_beta | Meta_se | Direction | Marginal p-Value | Conditional p-Value | Model R2 | Cross-Validation R2 | TWAS Reference Panel | Discovery Population |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ADAM15 | 1 | 155050566 | 155062775 | HCT | −0.078 | 0.018 | (−−−−+) | 8.74 | 2.52 | 0.301 | 0.395 | MESA | HL |
| THBS3 | 1 | 155195588 | 155209051 | HCT | 0.212 | 0.039 | (+++++) | 5.40 | 9.22 | 0.1 | 0.060 | MESA | HL |
| GTF2IRD2B | 7 | 75092573 | 75149817 | HCT | −0.249 | 0.056 | (−−−−−) | 9.18 | NA | 0.138 | 0.004 | MESA | HL |
| AGAP6 | 10 | 49982190 | 50010499 | HCT | −0.231 | 0.059 | (−−−−−) | 8.44 | NA | 0.119 | 0.017 | MESA | HL |
| SMAD6 | 15 | 66702228 | 66782849 | HCT | −0.356 | 0.081 | (−−−−+) | 1.14 | 8.55 | 0.074 | 0.005 | MESA | HL |
| ADAM15 | 1 | 155050566 | 155062775 | HGB | −0.066 | 0.018 | (−−−−+) | 3.95 | 1.51 | 0.301 | 0.395 | MESA | HL |
| THBS3 | 1 | 155195588 | 155209051 | HGB | 0.159 | 0.039 | (++++-) | 2.88 | 7.71 | 0.1 | 0.060 | MESA | HL |
| ARHGAP19 | 10 | 97222173 | 97292673 | HGB | −0.215 | 0.056 | (−−−−+) | 7.15 | NA | 0.1 | 0.011 | MESA | HL |
| CCDC15 | 11 | 124954121 | 125041489 | HGB | 0.059 | 0.016 | (++++−) | 5.84 | NA | 0.342 | 0.235 | MESA | HL |
| SMAD6 | 15 | 66702228 | 6678284 | HGB | −0.344 | 0.080 | (−−−−+) | 9.0 | 4.77 | 0.074 | 0.005 | MESA | HL |
| IL6R | 1 | 154405193 | 154469450 | PLT | −0.232 | 0.058 | (−−−−+) | 6.06 | 7.63 | 0.065 | 0.014 | MESA | HL |
| BAK1 | 6 | 33572547 | 33580293 | PLT | −0.118 | 0.029 | (−−−+−) | 4.95 | 2.42 | 0.167 | 0.088 | MESA | HL |
| PAQR8 | 6 | 52361421 | 52407777 | PLT | 0.080 | 0.019 | (+++++) | 4.79 | 1.27 | 0.268 | 0.165 | MESA | HL |
| TNFAIP2 | 14 | 103123442 | 103137439 | PLT | −0.265 | 0.065 | (−−−−−) | 6.85 | 8.10 | 0.095 | 0.013 | MESA | HL |
| SLC22A4 | 5 | 132294394 | 132344190 | WBC | 0.117 | 0.027 | (+++++) | 1.73 | 1.53 | 0.172 | 0.126 | MESA | HL |
| BAK1 | 6 | 33572547 | 33580293 | WBC | −0.110 | 0.028 | (−−−−+) | 9.47 | 1.46 | 0.167 | 0.088 | MESA | HL |
| GRINA | 8 | 143990056 | 143993415 | WBC | −0.298 | 0.077 | (−−+−−) | 9.70 | NA | 0.066 | 0.004 | MESA | HL |
| ATXN2 | 12 | 111443485 | 111599676 | WBC | −0.338 | 0.071 | (−−+−+) | 1.56 | 2.12 | 0.079 | 0.003 | MESA | HL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, J.; Xie, M.; Rowland, B.; Rosen, J.D.; Sun, Q.; Chen, J.; Tapia, A.L.; Qian, H.; Kowalski, M.H.; Shan, Y.; et al. Transcriptome-Wide Association Study of Blood Cell Traits in African Ancestry and Hispanic/Latino Populations. Genes 2021, 12, 1049. https://doi.org/10.3390/genes12071049
Wen J, Xie M, Rowland B, Rosen JD, Sun Q, Chen J, Tapia AL, Qian H, Kowalski MH, Shan Y, et al. Transcriptome-Wide Association Study of Blood Cell Traits in African Ancestry and Hispanic/Latino Populations. Genes. 2021; 12(7):1049. https://doi.org/10.3390/genes12071049
Chicago/Turabian StyleWen, Jia, Munan Xie, Bryce Rowland, Jonathan D. Rosen, Quan Sun, Jiawen Chen, Amanda L. Tapia, Huijun Qian, Madeline H. Kowalski, Yue Shan, and et al. 2021. "Transcriptome-Wide Association Study of Blood Cell Traits in African Ancestry and Hispanic/Latino Populations" Genes 12, no. 7: 1049. https://doi.org/10.3390/genes12071049
APA StyleWen, J., Xie, M., Rowland, B., Rosen, J. D., Sun, Q., Chen, J., Tapia, A. L., Qian, H., Kowalski, M. H., Shan, Y., Young, K. L., Graff, M., Argos, M., Avery, C. L., Bien, S. A., Buyske, S., Yin, J., Choquet, H., Fornage, M., ... Li, Y. (2021). Transcriptome-Wide Association Study of Blood Cell Traits in African Ancestry and Hispanic/Latino Populations. Genes, 12(7), 1049. https://doi.org/10.3390/genes12071049