
Non-Syndromic Autosomal Dominant Hearing Loss: The First Italian Family Carrying a Mutation in the NCOA3 Gene

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
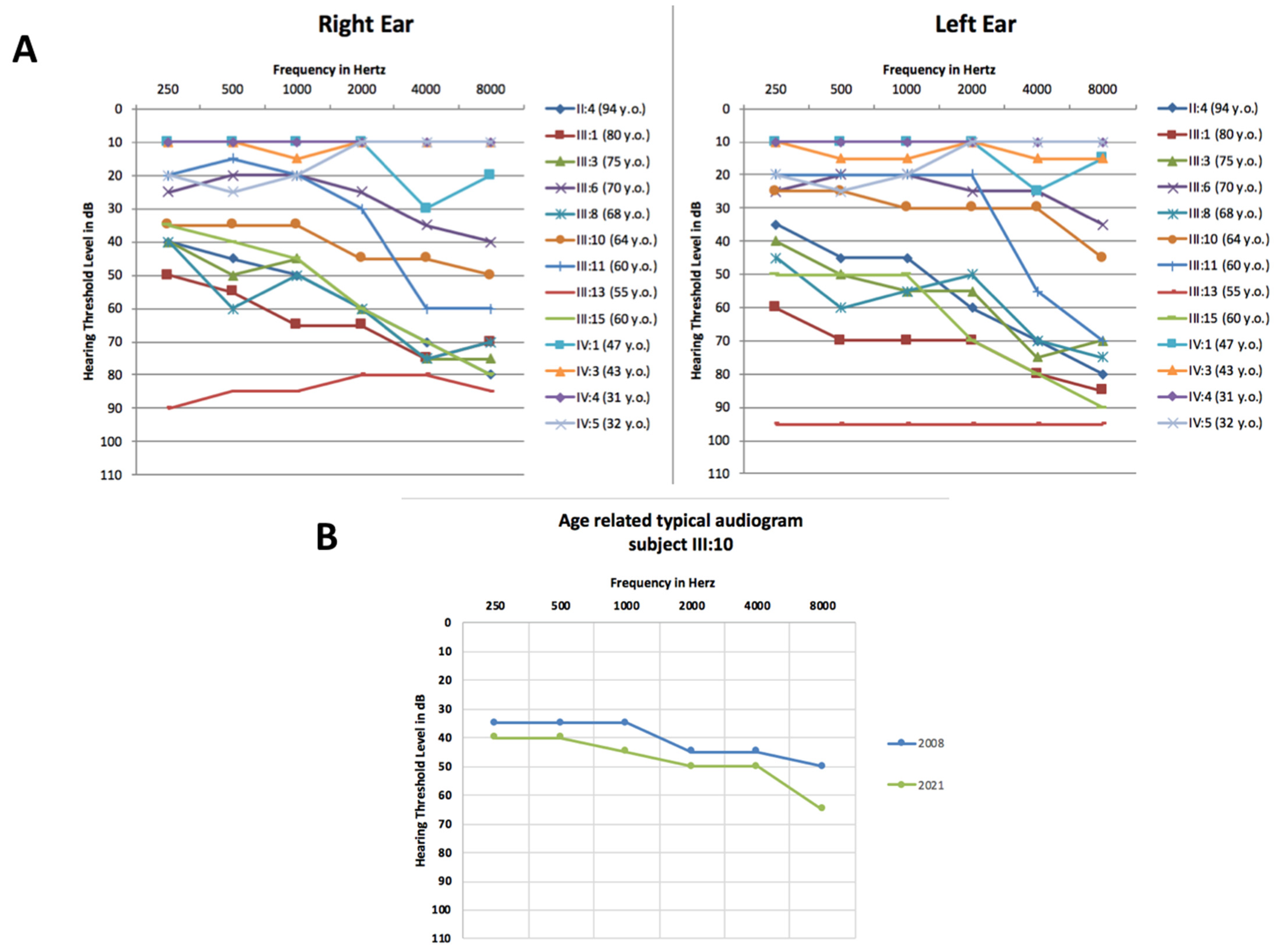
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Korver, A.M.; Smith, R.J.; Van Camp, G.; Schleiss, M.R.; Bitner-Glindzicz, M.A.; Lustig, L.R.; Usami, S.I.; Boudewyns, A.N. Congenital hearing loss. Nat. Rev. Dis. Primers 2017, 3, 16094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vona, B.; Doll, J.; Hofrichter, M.; Haaf, T. Non-syndromic hearing loss: Clinical and diagnostic challenges. Med. Genet. 2020, 32, 117–129. [Google Scholar] [CrossRef]
- Morgan, A.; Koboldt, D.C.; Barrie, E.S.; Crist, E.R.; García García, G.; Mezzavilla, M.; Faletra, F.; Mihalic Mosher, T.; Wilson, R.K.; Blanchet, C.; et al. Mutations in PLS1, encoding fimbrin, cause autosomal dominant nonsyndromic hearing loss. Hum. Mutat. 2019, 40, 2286–2295. [Google Scholar] [CrossRef]
- Sakuma, N.; Moteki, H.; Takahashi, M.; Nishio, S.Y.; Arai, Y.; Yamashita, Y.; Oridate, N.; Usami, S. An effective screening strategy for deafness in combination with a next-generation sequencing platform: A consecutive analysis. J. Hum. Genet. 2016, 61, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Sloan-Heggen, C.M.; Smith, R.J. Navigating genetic diagnostics in patients with hearing loss. Curr. Opin. Pediatr. 2016, 28, 705–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vona, B.; Nanda, I.; Hofrichter, M.A.; Shehata-Dieler, W.; Haaf, T. Non-syndromic hearing loss gene identification: A brief history and glimpse into the future. Mol. Cell. Probes 2015, 29, 260–270. [Google Scholar] [CrossRef] [PubMed]
- da Silva, R.S.; Dantas, V.; Alves, L.U.; Batissoco, A.C.; Oiticica, J.; Lawrence, E.A.; Kawafi, A.; Yang, Y.; Nicastro, F.S.; Novaes, B.C.; et al. NCOA3 identified as a new candidate to explain autosomal dominant progressive hearing loss. Hum. Mol. Genet. 2021, 29, 3691–3705. [Google Scholar] [CrossRef] [PubMed]
- Abecasis, G.R.; Cherny, S.S.; Cookson, W.O.; Cardon, L.R. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002, 30, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosmicki, J.A.; Samocha, K.E.; Howrigan, D.P.; Sanders, S.J.; Slowikowski, K.; Lek, M.; Karczewski, K.J.; Cutler, D.J.; Devlin, B.; Roeder, K.; et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet. 2017, 49, 504–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickerson, M.L.; Dancik, G.M.; Im, K.M.; Edwards, M.G.; Turan, S.; Brown, J.; Ruiz-Rodriguez, C.; Owens, C.; Costello, J.C.; Guo, G.; et al. Concurrent alterations in TERT, KDM6A, and the BRCA pathway in bladder cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 4935–4948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Ivanchenko, M.V.; Al Jandal, H.; Cicconet, M.; Indzhykulian, A.A.; Corey, D.P. PKHD1L1 is a coat protein of hair-cell stereocilia and is required for normal hearing. Nat. Commun. 2019, 10, 3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Limongelli, I.; Marini, S.; Bellazzi, R. PaPI: Pseudo amino acid composition to score human protein-coding variants. BMC Bioinform. 2015, 16, 123. [Google Scholar] [CrossRef] [Green Version]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [Green Version]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. ClinGen Hearing Loss Clinical Domain Working Group Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef]
- Yu, M.; Gilbert, S.; Li, Y.; Zhang, H.; Qiao, Y.; Lu, Y.; Tang, Y.; Zhen, Q.; Cheng, Y.; Liu, Y. Association of NCOA3 polymorphisms with Dyslipidemia in the Chinese Han population. Lipids Health Dis. 2015, 14, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Li, Z.; Wang, W.; Guo, J.; Kang, S.; Liu, S.; Li, H.; Wang, D.; Qi, X. NCOA3 Loss Disrupts Molecular Signature of Chondrocytes and Promotes Posttraumatic Osteoarthritis Progression. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 49, 2396–2413. [Google Scholar] [CrossRef]
- Sliwinska-Kowalska, M. Hearing. Handb. Clin. Neurol. 2015, 131, 341–363. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Mean Target Coverage (Mean Base Coverage Depth) | Coverage Uniformity (Percentage of Bases with a Coverage Depth > 0.2 × Mean_Coverage) | % Target Bases 10X (Percentage of Target Bases with Coverage above 10X) | |
|---|---|---|---|
| II:3 | 78.469824 | 97.5476 | 97.4332 |
| II:4 | 77.629201 | 95.2360 | 98.0183 |
| II:5 | 79.013458 | 96.1209 | 97.0431 |
| III:1 | 77.012397 | 98.0127 | 96.0126 |
| III:3 | 78.723768 | 97.5421 | 98.3218 |
| III:5 | 77.629071 | 96.9301 | 97.9854 |
| III:6 | 77.204720 | 95.9183 | 97.4510 |
| III:7 | 75.927492 | 96.5333 | 96.9981 |
| III:8 | 76.638206 | 98.1426 | 95.4832 |
| III:9 | 78.656329 | 95.6721 | 96.9453 |
| III:10 | 76.739260 | 98.2310 | 97.0034 |
| III:11 | 78.739279 | 96.9231 | 95.8741 |
| III:13 | 78.317429 | 97.8342 | 96.7632 |
| III:15 | 79.125820 | 96.7631 | 97.1632 |
| Subject | Affection | Variants | |||||
|---|---|---|---|---|---|---|---|
| ALDH1B1 NM_000692.5 | SMC2 NM_001042550.2 | NCO3 NM_181659.3 | MBD2 NM_015832.6 | KCNH6 NM_030779.4 | PKHD1L1 NM_177531.4 | ||
| c.G1087A | c.A1514G | c.G2909C | c.G707T | c.G263T | c.T9722A | ||
| II-3 (male, 92 y.o) | healthy | G/G | A/A | G/G | G/G | false positive | T/T |
| II-4 (female, 94 y.o.) | affected | G/A | A/G | G/C | G/T | false positive | T/A |
| II-5 (male, 88 y.o.) | affected | G/A | A/G | G/C | G/T | false positive | T/A |
| III-1 (male, 80 y.o.) | affected | G/A | A/G | G/C | G/T | false positive | T/T |
| III-3 (female, 75 y.o.) | affected | G/A | A/G | G/C | G/T | false positive | T/A |
| III-5 (male, 71 y.o.) | affected | G/G | A/A | G/C | G/G | false positive | T/T |
| III-6 (male, 70 y.o.) | affected | G/G | A/G | G/C | G/G | false positive | T/T |
| III-7 (female, 68 y.o.) | affected | G/A | A/G | G/C | G/G | false positive | T/T |
| III-8 (female, 70 y.o.) | affected | G/G | A/G | G/G | G/G | false positive | T/T |
| III-9 (male, 68 y.o.) | healthy | G/G | A/G | G/G | G/T | false positive | T/T |
| III-10 (male, 64 y.o.) | affected | G/A | A/G | G/C | G/T | false positive | T/A |
| III-11 (male, 60 y.o.) | affected | G/G | A/G | G/C | G/G | false positive | T/T |
| III-13 (female, 56 y.o.) | affected | G/A | A/G | G/C | G/T | false positive | T/A |
| III-15 (male, 60 y.o.) | affected | G/A | A/G | G/C | G/T | false positive | T/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tesolin, P.; Morgan, A.; Notarangelo, M.; Ortore, R.P.; Concas, M.P.; Notarangelo, A.; Girotto, G. Non-Syndromic Autosomal Dominant Hearing Loss: The First Italian Family Carrying a Mutation in the NCOA3 Gene. Genes 2021, 12, 1043. https://doi.org/10.3390/genes12071043
Tesolin P, Morgan A, Notarangelo M, Ortore RP, Concas MP, Notarangelo A, Girotto G. Non-Syndromic Autosomal Dominant Hearing Loss: The First Italian Family Carrying a Mutation in the NCOA3 Gene. Genes. 2021; 12(7):1043. https://doi.org/10.3390/genes12071043
Chicago/Turabian StyleTesolin, Paola, Anna Morgan, Michela Notarangelo, Rocco Pio Ortore, Maria Pina Concas, Angelantonio Notarangelo, and Giorgia Girotto. 2021. "Non-Syndromic Autosomal Dominant Hearing Loss: The First Italian Family Carrying a Mutation in the NCOA3 Gene" Genes 12, no. 7: 1043. https://doi.org/10.3390/genes12071043
APA StyleTesolin, P., Morgan, A., Notarangelo, M., Ortore, R. P., Concas, M. P., Notarangelo, A., & Girotto, G. (2021). Non-Syndromic Autosomal Dominant Hearing Loss: The First Italian Family Carrying a Mutation in the NCOA3 Gene. Genes, 12(7), 1043. https://doi.org/10.3390/genes12071043