
A Homozygous Synonymous Variant Likely Cause of Severe Ciliopathy Phenotype



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
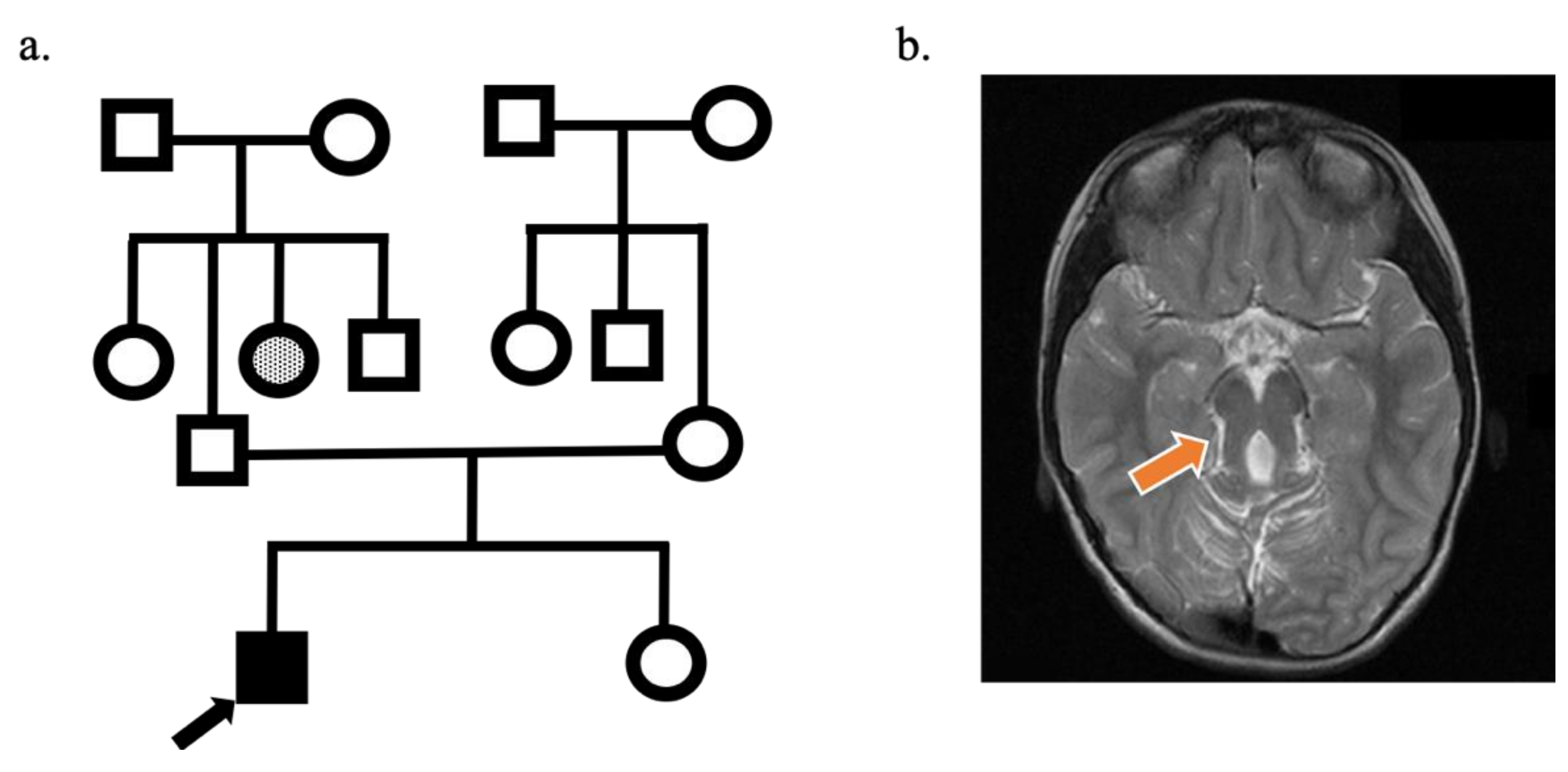
2.1. Subject
2.2. Molecular Genetic Analysis
3. Results
3.1. Neurological and Neuropsychiatric Findings
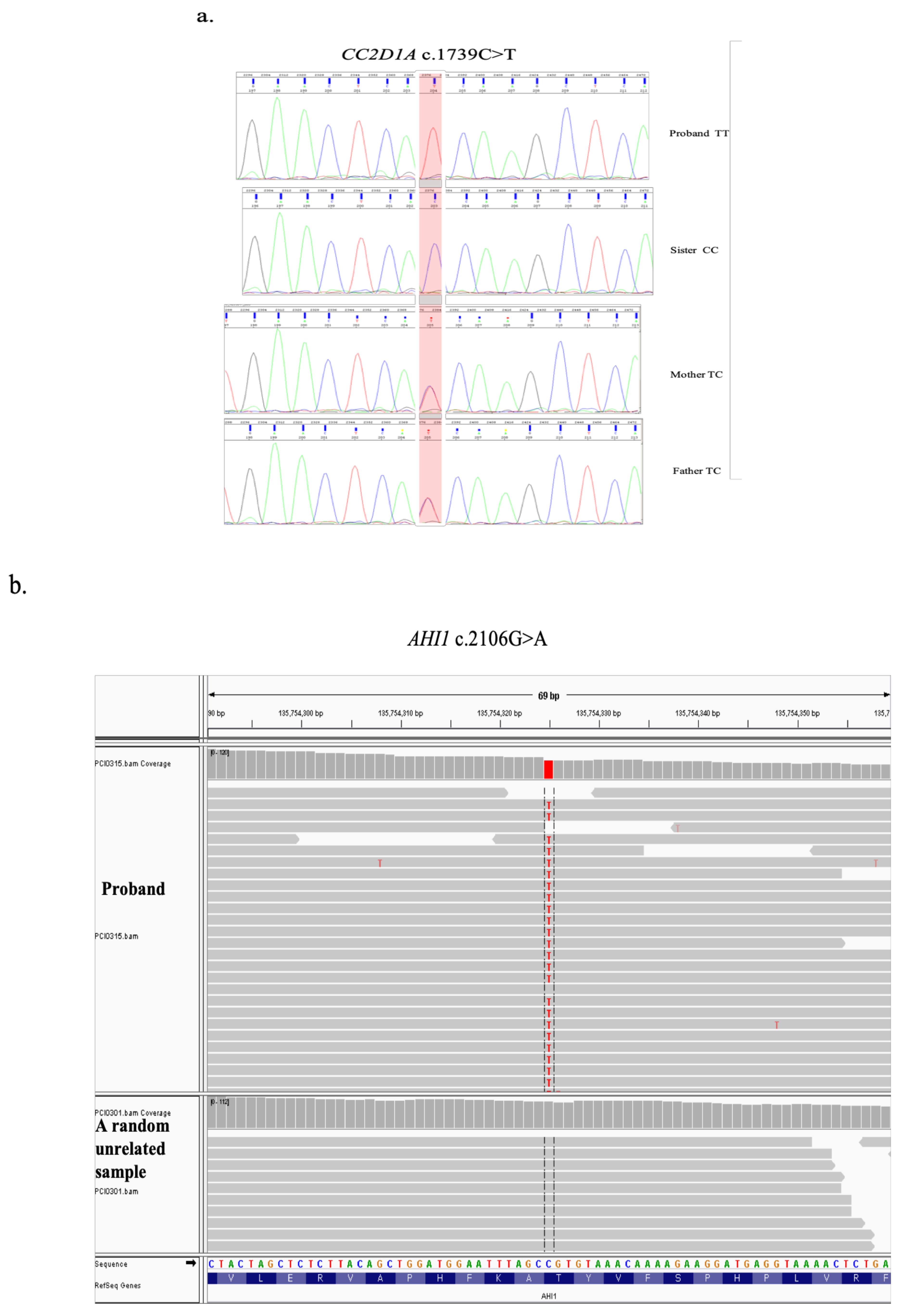
3.2. Molecular Genetic Findings
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Romani, M.; Micalizzi, A.; Valente, E.M. Joubert syndrome: Congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013, 12, 894–905. [Google Scholar] [CrossRef] [Green Version]
- Bachmann-Gagescu, R.; Dempsey, J.C.; Phelps, I.G.; O’Roak, B.J.; Knutzen, D.M.; Rue, T.C.; Ishak, G.E.; Isabella, C.R.; Gorden, N.; Adkins, J.; et al. Joubert syndrome: A model for untangling recessive disorders with extreme genetic heterogeneity. J. Med. Genet. 2015, 52, 514–522. [Google Scholar] [CrossRef] [Green Version]
- Edvardson, S.; Shaag, A.; Zenvirt, S.; Erlich, Y.; Hannon, G.J.; Shanske, A.L.; Gomori, J.M.; Ekstein, J.; Elpeleg, O. Joubert Syndrome 2 (JBTS2) in Ashkenazi Jews Is Associated with a TMEM216 Mutation. Am. J. Hum. Genet. 2010, 86, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Parisi, M.A.; Bennett, C.L.; Eckert, M.L.; Dobyns, W.B.; Gleeson, J.G.; Shaw, D.W.W.; McDonald, R.; Eddy, A.; Chance, P.F.; Glass, I.A. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am. J. Hum. Genet. 2004, 75, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Silhavy, J.L.; Brancati, F.; Barrano, G.; Krishnaswami, S.R.; Castori, M.; Lancaster, M.A.; Boltshauser, E.; Boccone, L.; Al-Gazali, L.; et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 2006, 38, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Otto, E.A.; Tory, K.; Attanasio, M.; Zhou, W.; Chaki, M.; Paruchuri, Y.; Wise, E.L.; Wolf, M.T.F.; Utsch, B.; Becker, C.; et al. Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J. Med. Genet. 2009, 46, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Delous, M.; Baala, L.; Salomon, R.; Laclef, C.; Vierkotten, J.; Tory, K.; Golzio, C.; Lacoste, T.; Besse, L.; Ozilou, C.; et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007, 39, 875–881. [Google Scholar] [CrossRef]
- Cantagrel, V.; Silhavy, J.L.; Bielas, S.L.; Swistun, D.; Marsh, S.E.; Bertrand, J.Y.; Audollent, S.; Attié-Bitach, T.; Holden, K.R.; Dobyns, W.B.; et al. Mutations in the Cilia Gene ARL13B Lead to the Classical Form of Joubert Syndrome. Am. J. Hum. Genet. 2008, 83, 170–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noor, A.; Windpassinger, C.; Patel, M.; Stachowiak, B.; Mikhailov, A.; Azam, M.; Irfan, M.; Siddiqui, Z.K.; Naeem, F.; Paterson, A.D.; et al. CC2D2A, Encoding A Coiled-Coil and C2 Domain Protein, Causes Autosomal-Recessive Mental Retardation with Retinitis Pigmentosa. Am. J. Hum. Genet. 2008, 82, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- Dafinger, C.; Liebau, M.C.; Elsayed, S.M.; Hellenbroich, Y.; Boltshauser, E.; Korenke, G.C.; Fabretti, F.; Janecke, A.R.; Ebermann, I.; Nürnberg, G.; et al. Mutations in KIF7 link Joubert syndrome with Sonic Hedgehog signaling and microtubule dynamics. J. Clin. Invest. 2011, 121, 2662–2667. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gonzalo, F.R.; Corbit, K.C.; Sirerol-Piquer, M.S.; Ramaswami, G.; Otto, E.A.; Noriega, T.R.; Seol, A.D.; Robinson, J.F.; Bennett, C.L.; Josifova, D.J.; et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 2011, 43, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Silhavy, J.L.; Lee, J.E.; Al-Gazali, L.; Thomas, S.; Davis, E.E.; Bielas, S.L.; Hill, K.J.; Iannicelli, M.; Brancati, F.; et al. Evolutionarily assembled cis-regulatory module at a human ciliopathy locus. Science 2012, 335, 966–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.E.; Silhavy, J.L.; Zaki, M.S.; Schroth, J.; Bielas, S.L.; Marsh, S.E.; Olvera, J.; Brancati, F.; Iannicelli, M.; Ikegami, K.; et al. CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat. Genet. 2012, 44, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srour, M.; Schwartzentruber, J.; Hamdan, F.F.; Ospina, L.H.; Patry, L.; Labuda, D.; Massicotte, C.; Dobrzeniecka, S.; Capo-Chichi, J.M.; Papillon-Cavanagh, S.; et al. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am. J. Hum. Genet. 2012, 90, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Legendre, M.; Saunier, S.; Bessières, B.; Alby, C.; Bonnière, M.; Toutain, A.; Loeuillet, L.; Szymanska, K.; Jossic, F.; et al. TCTN3 mutations cause Mohr-Majewski syndrome. Am. J. Hum. Genet. 2012, 91, 372–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaki, M.; Airik, R.; Ghosh, A.K.; Giles, R.H.; Chen, R.; Slaats, G.G.; Wang, H.; Hurd, T.W.; Zhou, W.; Cluckey, A.; et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012, 150, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Tuz, K.; Bachmann-Gagescu, R.; O’Day, D.R.; Hua, K.; Isabella, C.R.; Phelps, I.G.; Stolarski, A.E.; O’Roak, B.J.; Dempsey, J.C.; Lourenco, C.; et al. Mutations in CSPP1 cause primary cilia abnormalities and joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Am. J. Hum. Genet. 2014, 94, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Sang, L.; Miller, J.J.; Corbit, K.C.; Giles, R.H.; Brauer, M.J.; Otto, E.A.; Baye, L.M.; Wen, X.; Scales, S.J.; Kwong, M.; et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 2011, 145, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Srour, M.; Hamdan, F.F.; McKnight, D.; Davis, E.; Mandel, H.; Schwartzentruber, J.; Martin, B.; Patry, L.; Nassif, C.; Dionne-Laporte, A.; et al. Joubert syndrome in French Canadians and identification of mutations in CEP104. Am. J. Hum. Genet. 2015, 97, 744–753. [Google Scholar] [CrossRef] [Green Version]
- Sanders, A.A.W.M.; de Vrieze, E.; Alazami, A.M.; Alzahrani, F.; Malarkey, E.B.; Sorusch, N.; Tebbe, L.; Kuhns, S.; van Dam, T.J.P.; Alhashem, A.; et al. KIAA0556 is a novel ciliary basal body component mutated in Joubert syndrome. Genome Biol. 2015, 16. [Google Scholar] [CrossRef] [Green Version]
- Romani, M.; Micalizzi, A.; Kraoua, I.; Dotti, M.T.; Cavallin, M.; Sztriha, L.; Ruta, R.; Mancini, F.; Mazza, T.; Castellana, S.; et al. Mutations in B9D1 and MKS1 cause mild Joubert syndrome: Expanding the genetic overlap with the lethal ciliopathy Meckel syndrome. Orphanet J. Rare Dis. 2014, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Lambacher, N.J.; Bruel, A.L.; Van Dam, T.J.P.; Szymaska, K.; Slaats, G.G.; Kuhns, S.; McManus, G.J.; Kennedy, J.E.; Gaff, K.; Wu, K.M.; et al. TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nat. Cell Biol. 2016, 18, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Van De Weghe, J.C.; Rusterholz, T.D.S.; Latour, B.; Grout, M.E.; Aldinger, K.A.; Shaheen, R.; Dempsey, J.C.; Maddirevula, S.; Cheng, Y.H.H.; Phelps, I.G.; et al. Mutations in ARMC9, which Encodes a Basal Body Protein, Cause Joubert Syndrome in Humans and Ciliopathy Phenotypes in Zebrafish. Am. J. Hum. Genet. 2017, 101, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Davis, E.E.; Zhang, Q.; Liu, Q.; Diplas, B.H.; Davey, L.M.; Hartley, J.; Stoetzel, C.; Szymanska, K.; Ramaswami, G.; Logan, C.V.; et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011, 43, 189–196. [Google Scholar] [CrossRef]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VCV000461743.1—ClinVar—NCBI. 2018. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/461743/ (accessed on 7 June 2021).
- VCV000128618.11—ClinVar—NCBI. 2020. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/128618/ (accessed on 7 June 2021).
- Elhassanien, A.F.; Alghaiaty, H.A.A. Joubert syndrome: Clinical and radiological characteristics of nine patients. Ann. Indian Acad. Neurol. 2013, 16, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Incecik, F.; Hergüner, M.Ö.; Altunbaşak, Ş.; Gleeson, J.G. Joubert syndrome: Report of 11 cases. Turk. J. Pediatr. 2012, 54, 605–611. [Google Scholar]
- Brancati, F.; Dallapiccola, B.; Valente, E.M. Joubert Syndrome and related disorders. Orphanet J. Rare Dis. 2010, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Brancati, F.; Silhavy, J.L.; Castori, M.; Marsh, S.E.; Barrano, G.; Bertini, E.; Boltshauser, E.; Zaki, M.S.; Abdel-Aleem, A.; et al. AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann. Neurol. 2006, 59, 527–534. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Li, X.D.; Chen, Z. CC2D1A, a DM14 and C2 domain protein, activates NF-κB through the canonical pathway. J. Biol. Chem. 2010, 285, 24372–24380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, X.M.; Lemonde, S.; Jafar-Nejad, H.; Bown, C.D.; Goto, A.; Rogaeva, A.; Albert, P.R. Freud-1: A neuronal calcium-regulated repressor of the 5-HT1A receptor gene. J. Neurosci. 2003, 23, 7415–7425. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, A.; Arai, H.; Fujita, N. Centrosomal Aki1 and cohesin function in separase-regulated centriole disengagement. J. Cell Biol. 2009, 187, 607–614. [Google Scholar] [CrossRef]
- Basel-Vanagaite, L.; Attia, R.; Yahav, M.; Ferland, R.J.; Anteki, L.; Walsh, C.A.; Olender, T.; Straussberg, R.; Magal, N.; Taub, E.; et al. The CC2D1A, a member of a new gene family with C2 domains, is involved in autosomal recessive non-syndromic mental retardation. J. Med. Genet. 2006, 43, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Loviglio, M.N.; Beck, C.R.; White, J.J.; Leleu, M.; Harel, T.; Guex, N.; Niknejad, A.; Bi, W.; Chen, E.S.; Crespo, I.; et al. Identification of a RAI1-associated disease network through integration of exome sequencing, transcriptomics, and 3D genomics. Genome Med. 2016, 8, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shihab, H.A.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.M.; Gaunt, T.R. Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum. Genomics 2014, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Ma, A.C.H.; Mak, C.C.Y.; Yeung, K.S.; Pei, S.L.C.; Ying, D.; Yu, M.H.C.; Hasan, K.M.M.; Chen, X.; Chow, P.C.; Cheung, Y.F.; et al. Monoallelic Mutations in CC2D1A Suggest a Novel Role in Human Heterotaxy and Ciliary Dysfunction. Circ. Genomic Precis. Med. 2020, 13, 696–706. [Google Scholar] [CrossRef]
- Sakin, I.; Tuncel, G.; Sag, S.O.; Kaplan, O.; Khokha, M.; Ergoren, M.C.; Deniz, E.; Temel, S.G. CC2D1A as a novel ciliopathy gene. Eur. J. Human Genet. 2020, 28, 454. Available online: https://apps.webofknowledge.com/full_record.do?product=WOS&search_mode=GeneralSearch&qid=1&SID=D2vYSWWXyaCVjhBqCNA&page=1&doc=1 (accessed on 8 June 2021).
- Leitch, C.C.; Zaghloul, N.A.; Davis, E.E.; Stoetzel, C.; Diaz-Font, A.; Rix, S.; Al-Fadhel, M.; Lewis, R.A.; Eyaid, W.; Banin, E.; et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008, 40, 443–448. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuncel, G.; Kaymakamzade, B.; Engindereli, Y.; Temel, S.G.; Ergoren, M.C. A Homozygous Synonymous Variant Likely Cause of Severe Ciliopathy Phenotype. Genes 2021, 12, 945. https://doi.org/10.3390/genes12060945
Tuncel G, Kaymakamzade B, Engindereli Y, Temel SG, Ergoren MC. A Homozygous Synonymous Variant Likely Cause of Severe Ciliopathy Phenotype. Genes. 2021; 12(6):945. https://doi.org/10.3390/genes12060945
Chicago/Turabian StyleTuncel, Gulten, Bahar Kaymakamzade, Yeliz Engindereli, Sehime G. Temel, and Mahmut Cerkez Ergoren. 2021. "A Homozygous Synonymous Variant Likely Cause of Severe Ciliopathy Phenotype" Genes 12, no. 6: 945. https://doi.org/10.3390/genes12060945
APA StyleTuncel, G., Kaymakamzade, B., Engindereli, Y., Temel, S. G., & Ergoren, M. C. (2021). A Homozygous Synonymous Variant Likely Cause of Severe Ciliopathy Phenotype. Genes, 12(6), 945. https://doi.org/10.3390/genes12060945