
Models of Distal Arthrogryposis and Lethal Congenital Contracture Syndrome


Abstract
:1. Introduction
2. Muscle-Related Distal Arthrogryposis
2.1. MYH3
2.1.1. Biochemical and Cell Models for MYH3-Associated Distal Arthrogryposis
2.1.2. Invertebrate Models for MYH3-Associated Distal Arthrogryposis
2.1.3. Vertebrate Models for MYH3-Associated Distal Arthrogryposis
2.2. MYBPC1 and MYBPC2
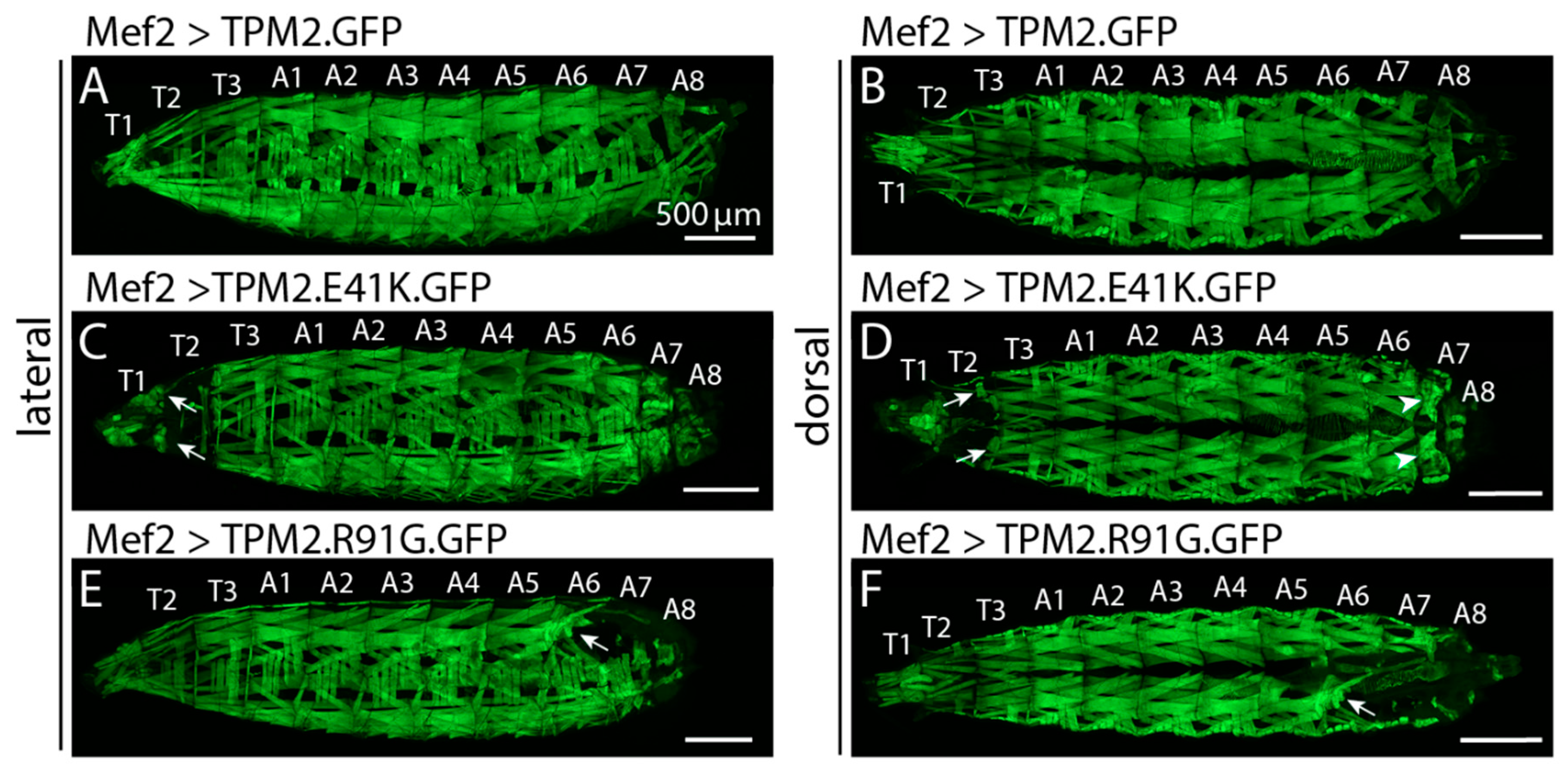
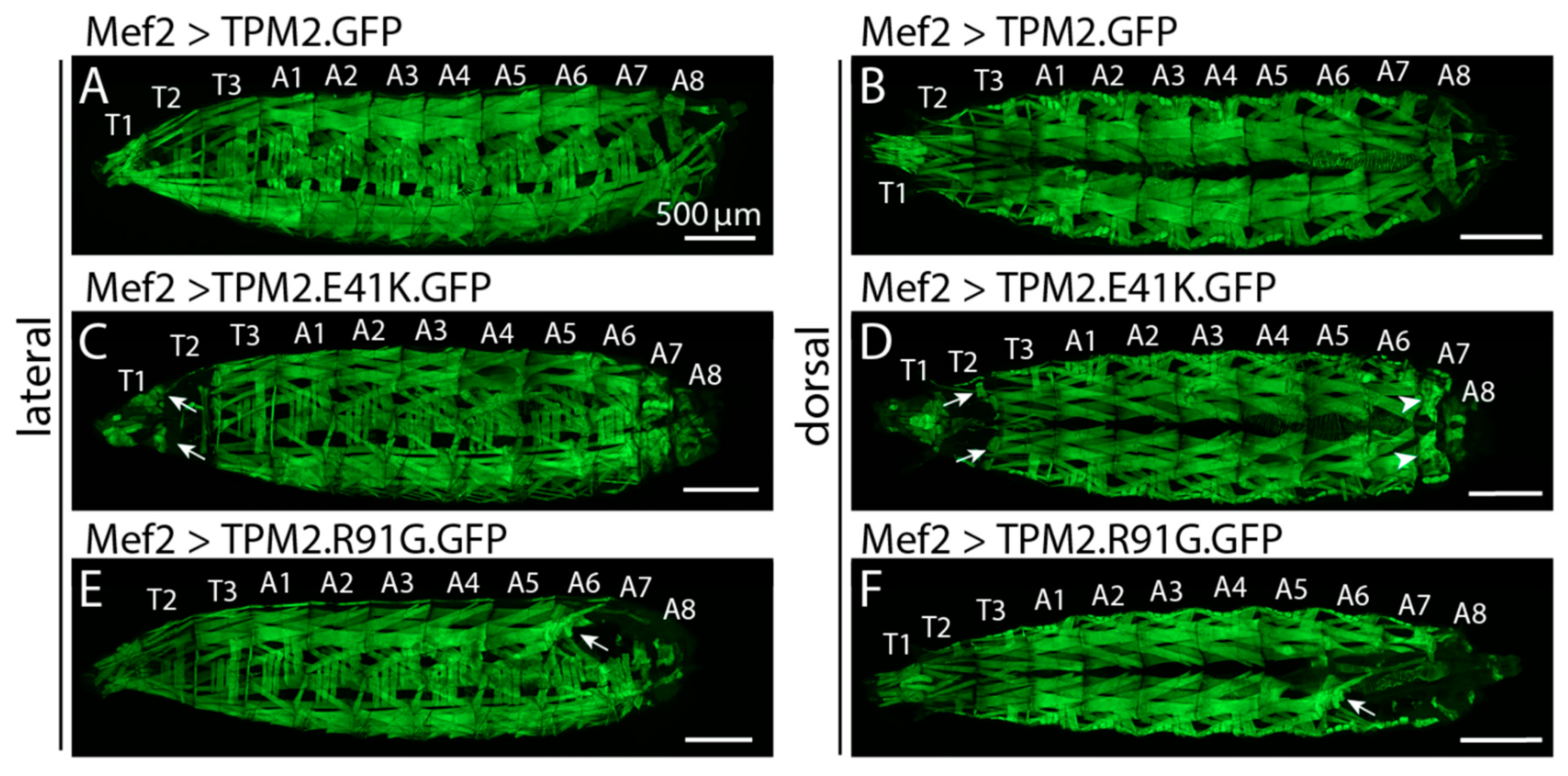
2.3. TPM2
2.4. TNNI2
2.5. TNNT3
2.6. MYLPF
3. Neural-Related Distal Arthrogryposis
3.1. PIEZO2
3.2. ECEL1
4. Lethal Congenital Contracture Syndrome
4.1. Nuclear mRNA Export (GLE1, ERBB3, and PIP5K1C)
4.2. Peripheral Nerve (CNTNAP1, ADGRG6, GLDN)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hall, J.G. Arthrogryposis (multiple congenital contractures): Diagnostic approach to etiology, classification, genetics, and general principles. Eur. J. Med. Genet. 2014, 57, 464–472. [Google Scholar] [CrossRef]
- Hall, J.G. Arthrogryposis multiplex congenita: Etiology, genetics, classification, diagnostic approach, and general aspects. J. Pediatric Orthop. 1997, 6, 159–166. [Google Scholar] [CrossRef]
- Ravenscroft, G.; Clayton, J.S.; Faiz, F.; Sivadorai, P.; Milnes, D.; Cincotta, R.; Moon, P.; Kamien, B.; Edwards, M.; Delatycki, M. Neurogenetic fetal akinesia and arthrogryposis: Genetics, expanding genotype-phenotypes and functional genomics. J. Med. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Felsenthal, N.; Zelzer, E. Mechanical regulation of musculoskeletal system development. Development 2017, 144, 4271–4283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiefer, J.; Hall, J.G. Gene ontology analysis of arthrogryposis (multiple congenital contractures). In American Journal of Medical Genetics Part C: Seminars in Medical Genetics; Wiley Online Library: Hoboken, NJ, USA, 2019; pp. 310–326. [Google Scholar]
- Bamshad, M.; Van Heest, A.E.; Pleasure, D. Arthrogryposis: A review and update. J. Bone. Jt. Surgery. Am. Vol. 2009, 91, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, A.E.; McMillin, M.J.; Gildersleeve, H.I.; Shively, K.; Tang, A.; Bamshad, M.J. Genotype-phenotype relationships in Freeman–Sheldon syndrome. Am. J. Med. Genet. Part A 2014, 164, 2808–2813. [Google Scholar] [CrossRef]
- Scala, M.; Accogli, A.; De Grandis, E.; Allegri, A.; Bagowski, C.P.; Shoukier, M.; Maghnie, M.; Capra, V. A novel pathogenic MYH3 mutation in a child with Sheldon–Hall syndrome and vertebral fusions. Am. J. Med. Genet. Part A 2018, 176, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Toydemir, R.M.; Rutherford, A.; Whitby, F.G.; Jorde, L.B.; Carey, J.C.; Bamshad, M.J. Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat. Genet. 2006, 38, 561–566. [Google Scholar] [CrossRef]
- Desai, D.; Stiene, D.; Song, T.; Sadayappan, S. Distal Arthrogryposis and Lethal Congenital Contracture Syndrome–An Overview. Front. Physiol. 2020, 11, 689. [Google Scholar] [CrossRef]
- Markus, B.; Narkis, G.; Landau, D.; Birk, R.Z.; Cohen, I.; Birk, O.S. Autosomal recessive lethal congenital contractural syndrome type 4 (LCCS4) caused by a mutation in MYBPC1. Hum. Mutat. 2012, 33, 1435–1438. [Google Scholar] [CrossRef]
- Racca, A.W.; Beck, A.E.; McMillin, M.J.; Korte, F.S.; Bamshad, M.J.; Regnier, M. The embryonic myosin R672C mutation that underlies Freeman-Sheldon syndrome impairs cross-bridge detachment and cycling in adult skeletal muscle. Hum. Mol. Genet. 2015, 24, 3348–3358. [Google Scholar] [CrossRef] [Green Version]
- Dieterich, K.; Le Tanno, P.; Kimber, E.; Jouk, P.S.; Hall, J.; Giampietro, P. The diagnostic workup in a patient with AMC: Overview of the clinical evaluation and paraclinical analyses with review of the literature. In American Journal of Medical Genetics Part C: Seminars in Medical Genetics; Wiley Online Library: Hoboken, NJ, USA, 2019; pp. 337–344. [Google Scholar]
- Guo, Y.; Kronert, W.A.; Hsu, K.H.; Huang, A.; Sarsoza, F.; Bell, K.M.; Suggs, J.A.; Swank, D.M.; Bernstein, S.I. Drosophila myosin mutants model the disparate severity of type 1 and type 2B distal arthrogryposis and indicate an enhanced actin affinity mechanism. Skelet. Muscle 2020, 10, 1–18. [Google Scholar] [CrossRef]
- Folkmann, A.W.; Dawson, T.R.; Wente, S.R. Insights into mRNA export-linked molecular mechanisms of human disease through a Gle1 structure–function analysis. Adv. Biol. Regul. 2014, 54, 74–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittle, J.; Antunes, L.; Harris, M.; Upshaw, Z.; Sepich, D.S.; Johnson, A.N.; Mokalled, M.; Solnica-Krezel, L.; Dobbs, M.B.; Gurnett, C.A. MYH 3-associated distal arthrogryposis zebrafish model is normalized with para-aminoblebbistatin. EMBO Mol. Med. 2020, 12, e12356. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, F.; Zhao, Y.; Yang, P.; Chen, J.; Sun, H.; Liu, L.; Li, W.; Pan, L.; Guo, Y. A gain-of-function mutation in Tnni2 impeded bone development through increasing Hif3a expression in DA2B mice. PLoS Genet. 2014, 10, e1004589. [Google Scholar] [CrossRef] [PubMed]
- Bayram, Y.; Karaca, E.; Akdemir, Z.C.; Yilmaz, E.O.; Tayfun, G.A.; Aydin, H.; Torun, D.; Bozdogan, S.T.; Gezdirici, A.; Isikay, S. Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J. Clin. Investig. 2016, 126, 762–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, M.A.; Rios, J.C.; Lu, Y.; Garcia-Fresco, G.P.; Ching, W.; Martin, M.S.; Li, J.; Einheber, S.; Chesler, M.; Rosenbluth, J. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron 2001, 30, 369–383. [Google Scholar] [CrossRef] [Green Version]
- Borovikov, Y.S.; Simonyan, A.O.; Karpicheva, O.E.; Avrova, S.V.; Rysev, N.A.; Sirenko, V.V.; Piers, A.; Redwood, C.S.J.B. The reason for a high Ca2+-sensitivity associated with Arg91Gly substitution in TPM2 gene is the abnormal behavior and high flexibility of tropomyosin during the ATPase cycle. Biochem. Biophys. Res. Commun. 2017, 494, 681–686. [Google Scholar] [CrossRef]
- Cameron-Christie, S.R.; Wells, C.F.; Simon, M.; Wessels, M.; Tang, C.Z.; Wei, W.; Takei, R.; Aarts-Tesselaar, C.; Sandaradura, S.; Sillence, D.O. Recessive Spondylocarpotarsal synostosis syndrome due to compound heterozygosity for variants in MYH3. Am. J. Hum. Genet. 2018, 102, 1115–1125. [Google Scholar] [CrossRef] [Green Version]
- Casey, J.P.; Brennan, K.; Scheidel, N.; McGettigan, P.; Lavin, P.T.; Carter, S.; Ennis, S.; Dorkins, H.; Ghali, N.; Blacque, O.E.; et al. Recessive NEK9 mutation causes a lethal skeletal dysplasia with evidence of cell cycle and ciliary defects. Hum. Mol. Genet. 2016, 25, 1824–1835. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.X.; Talbot, J.C.; Teets, E.M.; Previs, S.; Martin, B.L.; Shively, K.M.; Marvin, C.T.; Aylsworth, A.S.; Saadeh-Haddad, R.; Schatz, U.A. Mutations in MYLPF cause a novel segmental amyoplasia that manifests as distal arthrogryposis. Am. J. Hum. Genet. 2020, 107, 293–310. [Google Scholar] [CrossRef]
- Coste, B.; Houge, G.; Murray, M.F.; Stitziel, N.; Bandell, M.; Giovanni, M.A.; Philippakis, A.; Hoischen, A.; Riemer, G.; Steen, U. Gain-of-function mutations in the mechanically activated ion channel PIEZO2 cause a subtype of Distal Arthrogryposis. Proc. Natl. Acad. Sci. USA 2013, 110, 4667–4672. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Kumar, P.; Verma, A.; Maiti, T.K.; Mathew, S.J. Myosin heavy chain mutations that cause Freeman-Sheldon syndrome lead to muscle structural and functional defects in Drosophila. Dev. Biol. 2019, 449, 90–98. [Google Scholar] [CrossRef]
- Di Paolo, G.; Moskowitz, H.S.; Gipson, K.; Wenk, M.R.; Voronov, S.; Obayashi, M.; Flavell, R.; Fitzsimonds, R.M.; Ryan, T.A.; De Camilli, P.J.N. Impaired PtdIns (4, 5) P 2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 2004, 431, 415–422. [Google Scholar] [CrossRef]
- Durieux, A.-C.; Vignaud, A.; Prudhon, B.; Viou, M.T.; Beuvin, M.; Vassilopoulos, S.; Fraysse, B.; Ferry, A.; Lainé, J.; Romero, N.B.J.H.m.g. A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum. Mol. Genet. 2010, 19, 4820–4836. [Google Scholar] [CrossRef] [Green Version]
- Ekhilevitch, N.; Kurolap, A.; Oz-Levi, D.; Mory, A.; Hershkovitz, T.; Ast, G.; Mandel, H.; Baris, H. Expanding the MYBPC1 phenotypic spectrum: A novel homozygous mutation causes arthrogryposis multiplex congenita. Clin. Genet. 2016, 90, 84–89. [Google Scholar] [CrossRef]
- Folkmann, A.W.; Collier, S.E.; Zhan, X.; Ohi, M.D.; Wente, S.R. Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell 2013, 155, 582–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurnett, C.A.; Desruisseau, D.M.; McCall, K.; Choi, R.; Meyer, Z.I.; Talerico, M.; Miller, S.E.; Ju, J.-S.; Pestronk, A.; Connolly, A.M. Myosin binding protein C1: A novel gene for autosomal dominant distal arthrogryposis type 1. Hum. Mol. Genet. 2010, 19, 1165–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, K.; Buchan, J.G.; Alvarado, D.M.; Mccall, K.; Vydyanath, A.; Luther, P.K.; Goldsmith, M.I.; Dobbs, M.B.; Gurnett, C.A. MYBPC1 mutations impair skeletal muscle function in zebrafish models of arthrogryposis. Hum. Mol. Genet. 2013, 22, 4967–4977. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, M.; Sharma, A.; Kumar, P.; Kumar, A.; Bharadwaj, A.; Saini, M.; Kardon, G.; Mathew, S.J. Myosin heavy chain-embryonic regulates skeletal muscle differentiation during mammalian development. Development 2020, 147, dev184507. [Google Scholar] [CrossRef] [PubMed]
- Jao, L.-E.; Appel, B.; Wente, S.R. A zebrafish model of lethal congenital contracture syndrome 1 reveals Gle1 function in spinal neural precursor survival and motor axon arborization. Development 2012, 139, 1316–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, Y.; Li, J.; Xie, C.; Ritchlin, C.T.; Xing, L.; Hilton, M.J.; Schwarz, E.M. Troponin T3 expression in skeletal and smooth muscle is required for growth and postnatal survival: Characterization of Tnnt3tm2a (KOMP) Wtsi mice. Genesis 2013, 51, 667–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutsopoulos, O.S.; Kretz, C.; Weller, C.M.; Roux, A.; Mojzisova, H.; Böhm, J.; Koch, C.; Toussaint, A.; Heckel, E.; Stemkens, D.; et al. Dynamin 2 homozygous mutation in humans with a lethal congenital syndrome. Eur. J. Hum. Genet. 2013, 21, 637–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laquérriere, A.; Maluenda, J.; Camus, A.; Fontenas, L.; Dieterich, K.; Nolent, F.; Zhou, J.; Monnier, N.; Latour, P.; Gentil, D.; et al. Mutations in CNTNAP1 and ADCY6 are responsible for severe arthrogryposis multiplex congenita with axoglial defects. Hum. Mol. Genet. 2014, 23, 2279–2289. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Andersson-Lendahl, M.; Sejersen, T.; Arner, A. Knockdown of fast skeletal myosin-binding protein C in zebrafish results in a severe skeletal myopathy. J. Gen. Physiol. 2016, 147, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maluenda, J.; Manso, C.; Quevarec, L.; Vivanti, A.; Marguet, F.; Gonzales, M.; Guimiot, F.; Petit, F.; Toutain, A.; Whalen, S.; et al. Mutations in GLDN, encoding gliomedin, a critical component of the nodes of ranvier, are responsible for lethal arthrogryposis. Am. J. Hum. Genet. 2016, 99, 928–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyushenko, A.; Levitsky, D. Molecular mechanisms of pathologies of skeletal and cardiac muscles caused by point mutations in the tropomyosin genes. Biochemistry 2020, 85, 20–33. [Google Scholar] [CrossRef]
- McMillin, M.J.; Beck, A.E.; Chong, J.X.; Shively, K.M.; Buckingham, K.J.; Gildersleeve, H.I.; Aracena, M.I.; Aylsworth, A.S.; Bitoun, P.; Carey, J.C. Mutations in PIEZO2 cause Gordon syndrome, Marden-Walker syndrome, and distal arthrogryposis type 5. Am. J. Hum. Genet. 2014, 94, 734–744. [Google Scholar] [CrossRef] [Green Version]
- McMillin, M.J.; Below, J.E.; Shively, K.M.; Beck, A.E.; Gildersleeve, H.I.; Pinner, J.; Gogola, G.R.; Hecht, J.T.; Grange, D.K.; Harris, D.J. Mutations in ECEL1 cause distal arthrogryposis type 5D. Am. J. Hum. Genet. 2013, 92, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Monk, K.R.; Naylor, S.G.; Glenn, T.D.; Mercurio, S.; Perlin, J.R.; Dominguez, C.; Moens, C.B.; Talbot, W.S. AG protein–coupled receptor is essential for Schwann cells to initiate myelination. Science 2009, 325, 1402–1405. [Google Scholar] [CrossRef] [Green Version]
- Narkis, G.; Ofir, R.; Landau, D.; Manor, E.; Volokita, M.; Hershkowitz, R.; Elbedour, K.; Birk, O.S. Lethal contractural syndrome type 3 (LCCS3) is caused by a mutation in PIP5K1C, which encodes PIPKIγ of the phophatidylinsitol pathway. Am. J. Hum. Genet. 2007, 81, 530–539. [Google Scholar] [CrossRef] [Green Version]
- Narkis, G.; Ofir, R.; Manor, E.; Landau, D.; Elbedour, K.; Birk, O.S. Lethal congenital contractural syndrome type 2 (LCCS2) is caused by a mutation in ERBB3 (Her3), a modulator of the phosphatidylinositol-3-kinase/Akt pathway. Am. J. Hum. Genet. 2007, 81, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; Smith, L.L.; Faqeih, E.; Mohamed, J.; Gupta, V.A.; Alkuraya, F.S. ZBTB42 mutation defines a novel lethal congenital contracture syndrome (LCCS6). Hum. Mol. Genet. 2014, 23, 6584–6593. [Google Scholar] [CrossRef] [Green Version]
- Ravenscroft, G.; Nolent, F.; Rajagopalan, S.; Meireles, A.M.; Paavola, K.J.; Gaillard, D.; Alanio, E.; Buckland, M.; Arbuckle, S.; Krivanek, M.; et al. Mutations of GPR126 are responsible for severe arthrogryposis multiplex congenita. Am. J. Hum. Genet. 2015, 96, 955–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riethmacher, D.; Sonnenberg-Riethmacher, E.; Brinkmann, V.; Yamaai, T.; Lewin, G.R.; Birchmeier, C. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature 1997, 389, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Sandaradura, S.A.; Bournazos, A.; Mallawaarachchi, A.; Cummings, B.B.; Waddell, L.B.; Jones, K.J.; Troedson, C.; Sudarsanam, A.; Nash, B.M.; Peters, G.B.; et al. Nemaline myopathy and distal arthrogryposis associated with an autosomal recessive TNNT3 splice variant. Hum. Mutat. 2018, 39, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shashi, V.; Geist, J.; Lee, Y.; Yoo, Y.; Shin, U.; Schoch, K.; Sullivan, J.; Stong, N.; Smith, E.; Jasien, J. Heterozygous variants in MYBPC1 are associated with an expanded neuromuscular phenotype beyond arthrogryposis. Hum. Mutat. 2019, 40, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.S.; Brassington, A.-M.E.; Grannatt, K.; Rutherford, A.; Whitby, F.G.; Krakowiak, P.A.; Jorde, L.B.; Carey, J.C.; Bamshad, M. Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes. Am. J. Hum. Genetics 2003, 72, 681–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toydemir, R.M.; Chen, H.; Proud, V.K.; Martin, R.; van Bokhoven, H.; Hamel, B.C.; Tuerlings, J.H.; Stratakis, C.A.; Jorde, L.B.; Bamshad, M.J. Trismus-pseudocamptodactyly syndrome is caused by recurrent mutation of MYH8. Am. J. Med Genet. Part A 2006, 140, 2387–2393. [Google Scholar] [CrossRef] [PubMed]
- Veugelers, M.; Bressan, M.; McDermott, D.A.; Weremowicz, S.; Morton, C.C.; Mabry, C.C.; Lefaivre, J.-F.; Zunamon, A.; Destree, A.; Chaudron, J.-M.; et al. Mutation of perinatal myosin heavy chain associated with a Carney complex variant. N. Engl. J. Med. 2004, 351, 460–469. [Google Scholar] [CrossRef]
- Vigoreaux, J.O. Genetics of the Drosophila flight muscle myofibril: A window into the biology of complex systems. Bioessays 2001, 23, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Walklate, J.; Vera, C.; Bloemink, M.J.; Geeves, M.A.; Leinwand, L. The most prevalent Freeman-Sheldon Syndrome mutations in the embryonic myosin motor share functional defects. J. Biol. Chem. 2016, 291, 10318–10331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhou, H.; Zhang, M.; Liu, W.; Deng, T.; Zhao, Q.; Li, Y.; Lei, J.; Li, X.; Xiao, B. Structure and mechanogating of the mammalian tactile channel PIEZO2. Nature 2019, 573, 225–229. [Google Scholar] [CrossRef]
- Williams, J.; Boin, N.G.; Valera, J.M.; Johnson, A.N. Noncanonical roles for Tropomyosin during myogenesis. Development 2015, 142, 3440–3452. [Google Scholar] [PubMed] [Green Version]
- Chong, J.X.; Burrage, L.C.; Beck, A.E.; Marvin, C.T.; McMillin, M.J.; Shively, K.M.; Harrell, T.M.; Buckingham, K.J.; Bacino, C.A.; Jain, M.; et al. Autosomal-dominant multiple pterygium syndrome is caused by mutations in MYH3. Am. J. Hum. Genet. 2015, 96, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhong, B.; Han, W.; Zhao, N.; Liu, W.; Sui, Y.; Wang, Y.; Lu, Y.; Wang, H.; Li, J. Two novel mutations in myosin binding protein C slow causing distal arthrogryposis type 2 in two large Han Chinese families may suggest important functional role of immunoglobulin domain C2. PLoS ONE 2015, 10, e0117158. [Google Scholar] [CrossRef] [Green Version]
- Alvarado, D.M.; Buchan, J.G.; Gurnett, C.A.; Dobbs, M.B. Exome sequencing identifies an MYH3 mutation in a family with distal arthrogryposis type 1. J. Bone Jt. Surg. Am. Vol. 2011, 93, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, A.E.; McMillin, M.J.; Gildersleeve, H.I.; Kezele, P.R.; Shively, K.M.; Carey, J.C.; Regnier, M.; Bamshad, M.J. Spectrum of mutations that cause distal arthrogryposis types 1 and 2B. Am. J. Med. Genet. Part A 2013, 161, 550–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zieba, J.; Zhang, W.; Chong, J.X.; Forlenza, K.N.; Martin, J.H.; Heard, K.; Grange, D.K.; Butler, M.G.; Kleefstra, T.; Lachman, R.S. A postnatal role for embryonic myosin revealed by MYH3 mutations that alter TGFβ signaling and cause autosomal dominant spondylocarpotarsal synostosis. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gil-Gálvez, A.; Carbonell-Corvillo, P.; Paradas, C.; Miranda-Vizuete, A. Cautionary note on the use of Caenorhabditis elegans to study muscle phenotypes caused by mutations in the human MYH7 gene. BioTechniques 2020, 68, 296–299. [Google Scholar] [CrossRef] [Green Version]
- Repetti, G.G.; Toepfer, C.N.; Seidman, J.G.; Seidman, C.E. Novel therapies for prevention and early treatment of cardiomyopathies: Now and in the future. Circ. Res. 2019, 124, 1536–1550. [Google Scholar] [CrossRef]
- Tajsharghi, H.; Ohlsson, M.; Palm, L.; Oldfors, A. Myopathies associated with β-tropomyosin mutations. Neuromuscul. Disord. 2012, 22, 923–933. [Google Scholar] [CrossRef]
- Monnier, N.; Lunardi, J.; Marty, I.; Mezin, P.; Labarre-Vila, A.; Dieterich, K.; Jouk, P.S. Absence of β-tropomyosin is a new cause of Escobar syndrome associated with nemaline myopathy. Neuromuscul. Disord. 2009, 19, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Szczesna-Cordary, D.; Craig, R.; Diaz-Perez, Z.; Guzman, G.; Miller, T.; Potter, J.D. Fast skeletal muscle regulatory light chain is required for fast and slow skeletal muscle development. FASEB J. 2007, 21, 2205–2214. [Google Scholar] [CrossRef]
- Assaraf, E.; Blecher, R.; Heinemann-Yerushalmi, L.; Krief, S.; Vinestock, R.C.; Biton, I.E.; Brumfeld, V.; Rotkopf, R.; Avisar, E.; Agar, G.; et al. Piezo2 expressed in proprioceptive neurons is essential for skeletal integrity. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, A.; Valdenaire, O.; Köster, A.; Lang, Y.; Schmitt, G.; Lenz, B.; Bluethmann, H.; Rohrer, J. Neonatal lethality in mice deficient in XCE, a novel member of the endothelin-converting enzyme and neutral endopeptidase family. J. Biol. Chem. 1999, 274, 20450–20456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, N.V.; Brueton, L.A.; Cox, P.; Greally, M.T.; Tolmie, J.; Pasha, S.; Aligianis, I.A.; van Bokhoven, H.; Marton, T.; Al-Gazali, L. Mutations in the embryonal subunit of the acetylcholine receptor (CHRNG) cause lethal and Escobar variants of multiple pterygium syndrome. Am. J. Hum. Genet. 2006, 79, 390–395. [Google Scholar] [CrossRef] [Green Version]
- Nousiainen, H.O.; Kestilä, M.; Pakkasjärvi, N.; Honkala, H.; Kuure, S.; Tallila, J.; Vuopala, K.; Ignatius, J.; Herva, R.; Peltonen, L. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat. Genet. 2008, 40, 155–157. [Google Scholar] [CrossRef]
- Eshed, Y.; Feinberg, K.; Poliak, S.; Sabanay, H.; Sarig-Nadir, O.; Spiegel, I.; Bermingham, J.R., Jr.; Peles, E. Gliomedin mediates Schwann cell-axon interaction and the molecular assembly of the nodes of Ranvier. Neuron 2005, 47, 215–229. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Gene | Full Name | Disorder | Inheritance Pattern | Modeled in | Source Human | Models of Disease Source |
|---|---|---|---|---|---|---|
| ARTHROGRYPOSIS | ||||||
| MYH3 | Myosin, Heavy Polypeptide 3, Skeletal Muscle, Embryonic | DA1, DA2A, DA2B, DA8, Spondylocarpotarsal Syndrome | AD, AR | Zebrafish, Cell, Biochemical | Toydemir et al., 2006b [9]; Chong et al., 2015 [57]; Cameron-Christie et al., 2019 [21] | Racca et al., 2015 [12]; Walklate et al., 2016 [54]; Wang et al., 2019 [55]; Whittle et al., 2020 [16]; Guo et al., 2020 [14]; Das et al., 2019 [25] |
| TPM2 | Tropomyosin 2 | DA1, Cap Myopathy, Nemaline Myopathy | AD, AR | Drosophila, Biochemical | Sung et al., 2003 [50] | Williams et al., 2015 [56]; Borovikov et al., 2017 [20]; Matyushenko & Levitsky, 2020 [39]; |
| MYLPF | Myosin Regulatory Light Chain 2, Skeletal Muscle Isoform | DA1, DA2B | AD, AR | Zebrafish | Chong et al., 2020 [23] | Chong et al., 2020 [23] |
| MYBPC1 | Myosin-Binding Protein C, Slow-Type | DA1, DA2, LCCS4 | AD | Zebrafish | Gurnett et al., 2010 [30]; Li et al., 2015 [58]; Ekhilevitch et al., 2016 [28]; Shashi et al., 2019 [49]; | Ha et al., 2013 [31] |
| MYBPC2 | Myosin-Binding Protein C, Fast-Type | DA (unspecified) | AD | Zebrafish | Bayram et al., 2016 [18] | Li et al., 2016 [37] |
| TNNT3 | Troponin T3, Fast Skeletal Type | DA2B | AD, AR | Mouse | Sung et al., 2003 [50]; Sandaradura et al., 2018 [48] | Ju et al., 2013 [34] |
| TNNI2 | Troponin I2, Fast Skeletal Type | DA2B | AD | Mouse, Drosophila | Sung et al., 2003 [50] | Zhu et al., 2014 [17]; Vigoreaux, 2001 [53] |
| PIEZO2 | Piezo Type Mechanosensitive Ion Channel Component 2 | DA3, DA5 | AR | Cell | McMillin et al., 2014 [40] | Coste et al., 2013 [24]; McMillin et al., 2014 [40] |
| ECEL1 | Endothelin Converting Enzyme Like 1 | DA5 (or DA5D) | AR | - | McMillin et al., 2013 [41] | - |
| MYH8 | Myosin, Heavy Polypeptide 8, Skeletal Muscle, Fetal | DA7 | AD | - | Toydemir et al., 2006a; [51] Veugelers et al., 2004 [52] | - |
| LETHAL CONGENITAL CONTRATURE SYNDROME | ||||||
| GLE1 | GLE1 RNA Export Mediator | LCCS1 | AR | Zebrafish, Cell, Biochemical | Jao et al., 2012 [33] | Folkmann et al., 2013 [29]; Jao et al., 2012 [33] |
| ERBB3 | ERB-B2 Receptor Tyrosine Kinase 3 | LCCS2 | AR | Mouse | Narkis et al., 2007 [44] | Riethmacher et al., 1997 [47] |
| PIP5K1C | Phosphatidylinositol 4-Phosphate 5-Kinase, type 1, gamma | LCCS3 | AR | Mouse | Narkis et al., 2007 [43] | DiPaolo et al., 2004 [26] |
| MYBPC1 | Myosin-Binding Protein C, Slow-Type | LCCS4, DA1, DA2 | AD, AR | Zebrafish | Markus et al., 2012 [11] | Ha et al., 2013 [31] |
| DNM2 | Dynamin, 2 | LCCS5, Centronuclear Myopathy, CMT2M, CMT Intermed | AD, AR | Mouse | Koutsopoulos et al., 2013 [35] | Durieux et al., 2010 [27]; Koutsopoulos et al., 2013 [35] |
| ZBTB42 | Zinc finger-and BTB Domain-containing Protein 42 | LCCS6 | AR | Zebrafish | Patel et al., 2014 [45] | Patel et al., 2014 [45] |
| CNTNAP1 | Contactin-associated protein 1 | LCCS7, Congenital Hypomyelinating Neuropathy | AR | Mouse | Laquerriere et al., 2014 [36] | Bhat et al., 2001 [19] |
| ADCY6 | Adenylyl cyclase 6 | LCCS8 | AR | Zebrafish | Laquerriere et al., 2014 [36] | Laquerriere et al., 2014 [36] |
| ADGRG6 | Adhesion G-protein coupled receptor G6 or GPR126 | LCCS9 | AR | Zebrafish | Ravenscroft et al., 2015 [46] | Monk et al., 2009 [42] |
| NEK9 | Nima-related kinase 1 | LCCS10 | AR | - | Casey et al., 2016 [22] | - |
| GLDN | Gliomedin | LCCS11 | AR | - | Maluenda et al., 2016 [38] | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whittle, J.; Johnson, A.; Dobbs, M.B.; Gurnett, C.A. Models of Distal Arthrogryposis and Lethal Congenital Contracture Syndrome. Genes 2021, 12, 943. https://doi.org/10.3390/genes12060943
Whittle J, Johnson A, Dobbs MB, Gurnett CA. Models of Distal Arthrogryposis and Lethal Congenital Contracture Syndrome. Genes. 2021; 12(6):943. https://doi.org/10.3390/genes12060943
Chicago/Turabian StyleWhittle, Julia, Aaron Johnson, Matthew B. Dobbs, and Christina A. Gurnett. 2021. "Models of Distal Arthrogryposis and Lethal Congenital Contracture Syndrome" Genes 12, no. 6: 943. https://doi.org/10.3390/genes12060943
APA StyleWhittle, J., Johnson, A., Dobbs, M. B., & Gurnett, C. A. (2021). Models of Distal Arthrogryposis and Lethal Congenital Contracture Syndrome. Genes, 12(6), 943. https://doi.org/10.3390/genes12060943