
Cis-Segregation of c.1171C>T Stop Codon (p.R391*) in SERPINC1 Gene and c.1691G>A Transition (p.R506Q) in F5 Gene and Selected GWAS Multilocus Approach in Inherited Thrombophilia

 ,
,  , ,
, ,
Abstract
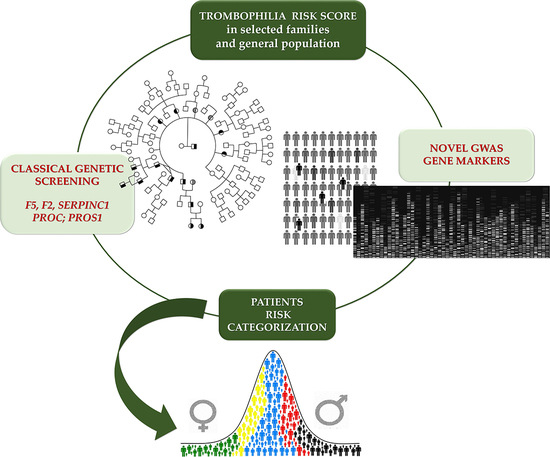
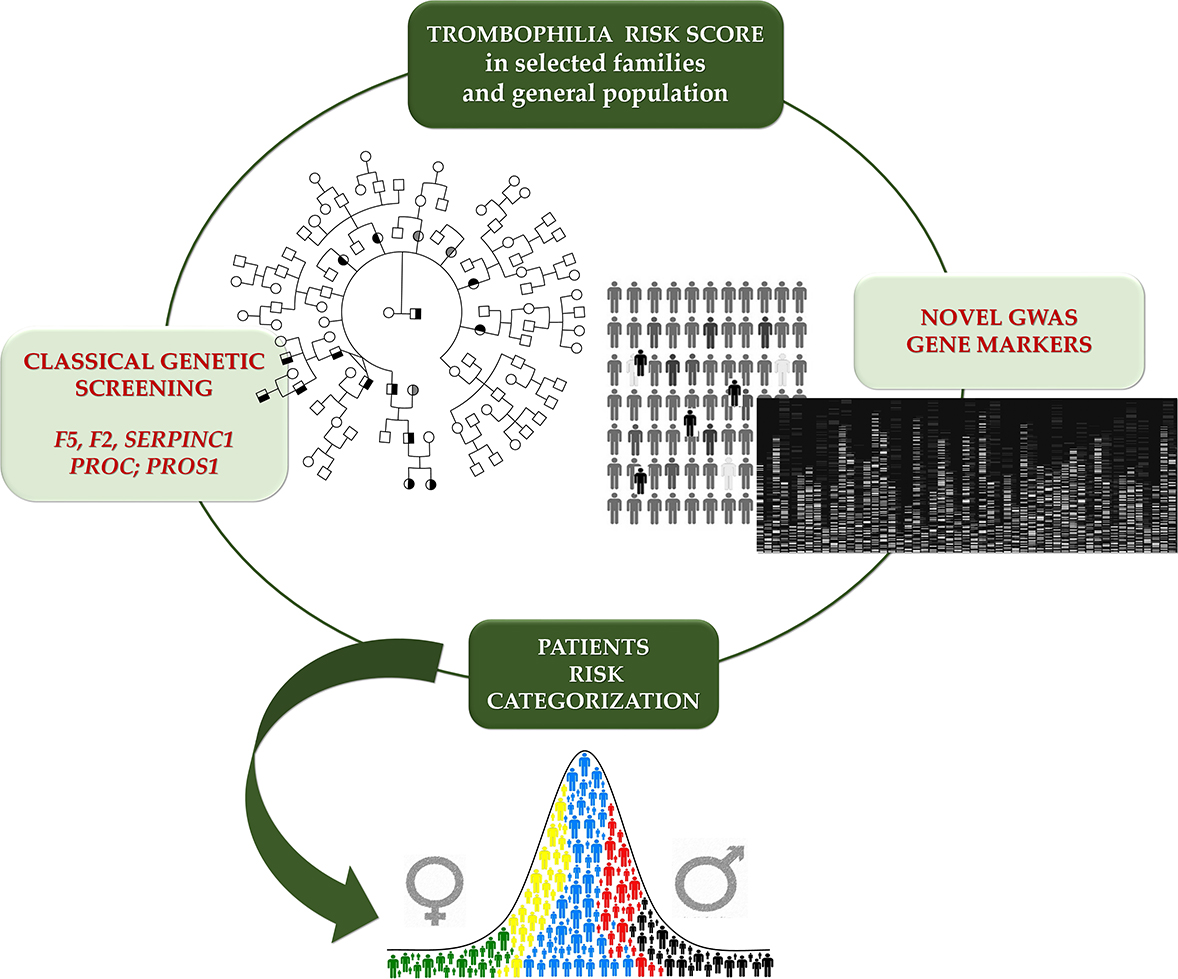
1. Introduction
2. Material and Methods
2.1. Family and Study Design
2.2. DNA Extraction, PCR Conditions, and Sequencing
2.3. STRs Linkage Analyses
2.4. Restriction Analyses
3. Results
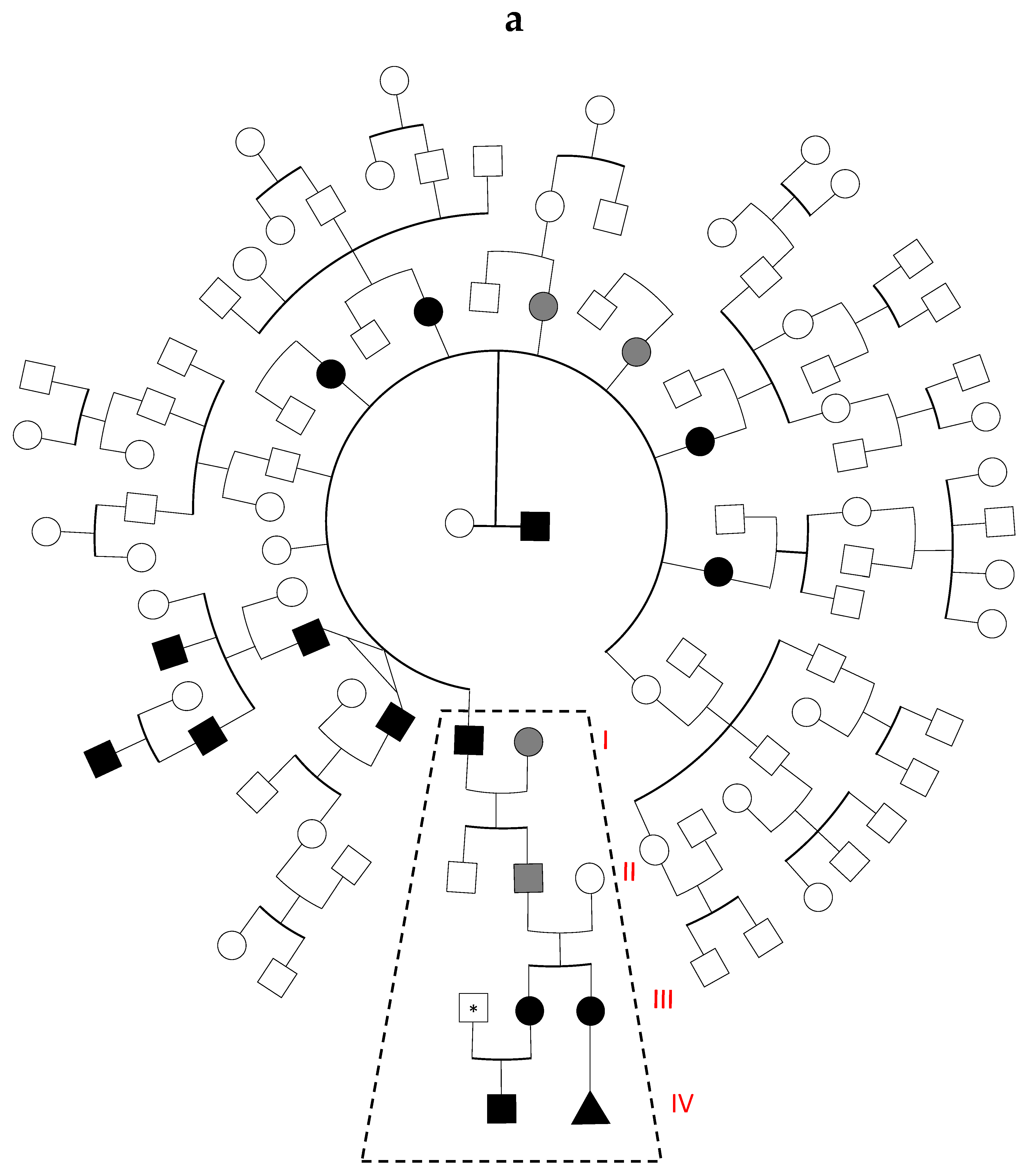
3.1. Family History and Index Cases
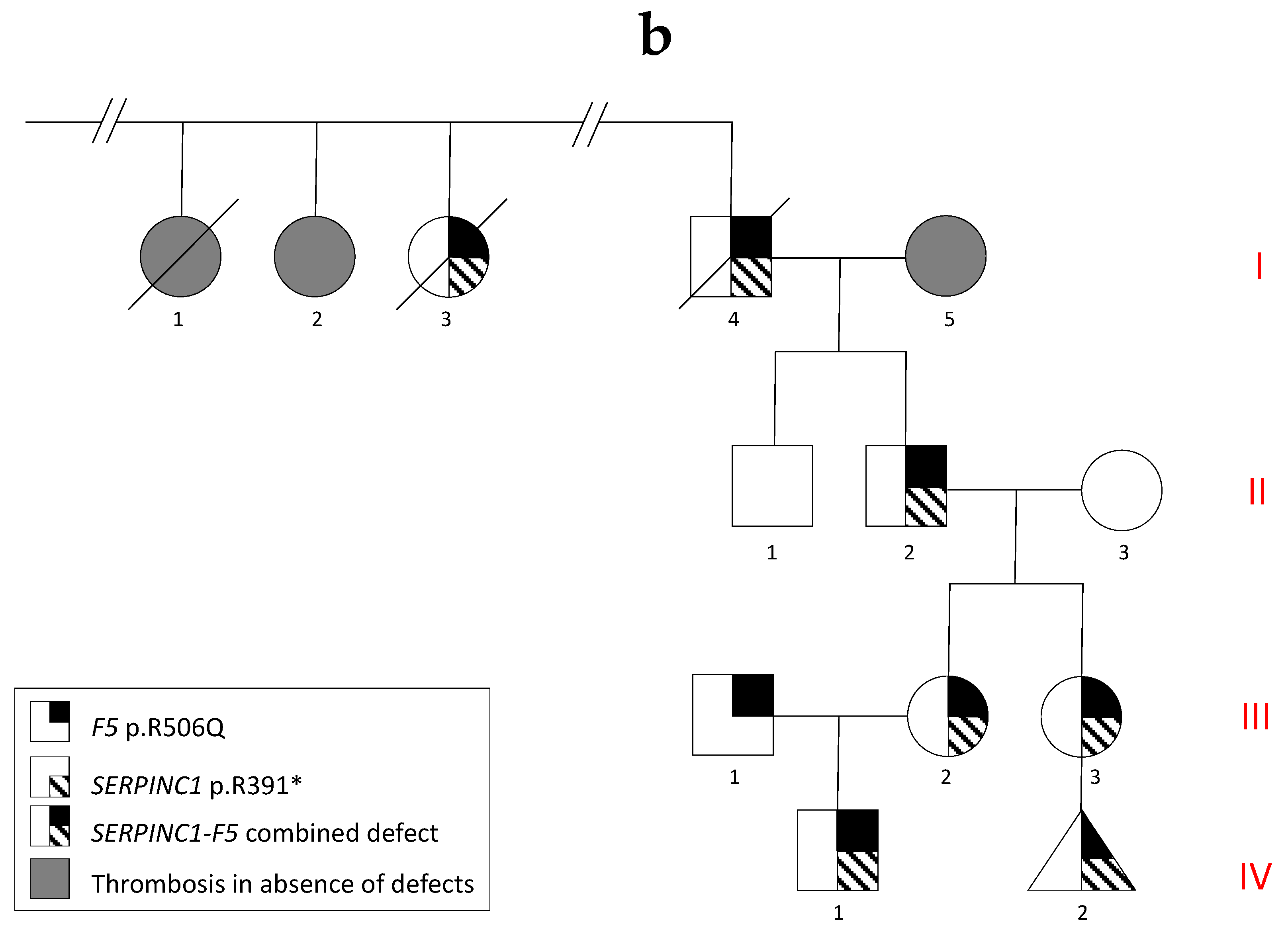
3.2. Genetic Analyses
3.3. Family Linkage Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Boven, H.H.; Reitsma, P.H.; Rosendaal, F.R.; Bayston, T.A.; Chowdhury, V.; Bauer, K.A.; Scharrer, I.; Conard, J.; Lane, D.A. Factor V Leiden (FV R506Q) in families with inherited antithrombin deficiency. Thromb. Haemost. 1996, 75, 417–421. [Google Scholar] [CrossRef]
- Gemmati, D.; Serino, M.L.; Moratelli, S.; Mari, R.; Ballerini, G.; Scapoli, G.L. Coexistence of antithrombin deficiency, factor V Leiden and hyperhomocysteinemia in a thrombotic family. Blood Coagul. Fibrinolysis 1998, 9, 173–176. [Google Scholar] [CrossRef]
- Ordonez, A.; de Cos, C.; Minano, A.; Rodriguez, J.; Hernandez-Espinosa, D.; Munoz, J.A.; Gonzalez-Conejero, R.; Vicente, V.; Corral, J. Coexistence of three genetic risk factors in a Spanish thrombophilic family: Factor V Leiden, prothrombin 20210 and a new type I antithrombin deficiency. Thromb. Haemost. 2007, 97, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Serino, M.L.; Tognazzo, S.; Ongaro, A.; Moratelli, S.; Gilli, G.; Forini, E.; De Mattei, M.; Scapoli, G.L. The reduced sensitivity of the ProC Global test in protein S deficient subjects reflects a reduction in the associated thrombotic risk. Blood Coagul. Fibrinolysis 2001, 12, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Serino, M.L.; Moratelli, S.; Tognazzo, S.; Ongaro, A.; Scapoli, G.L. Coexistence of factor V G1691A and factor II G20210A gene mutations in a thrombotic family is associated with recurrence and early onset of venous thrombosis. Haemostasis 2001, 31, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Serino, M.L.; Scapoli, G.L. A modified functional global test to measure protein C, protein S activities and the activated protein C-resistance phenotype. Thromb. Res. 1998, 92, 141–148. [Google Scholar] [CrossRef]
- Gemmati, D.; Serino, M.L.; Verzola, I.; Mari, R.; Moratelli, S.; Ballerini, G. Resistance to activated protein C and low levels of protein S activity in nine thrombophilic families: A correct diagnosis. Blood Coagul. Fibrinolysis 1997, 8, 118–123. [Google Scholar] [CrossRef]
- Gemmati, D.; Serino, M.L.; Mari, R.; Verzola, I.; Moratelli, S.; Ballerini, G. Different Anticoagulant Response to Activated Protein C (APC test) and to Agkistrodon Contortix Venom (ACV test) in a Family with FV-R506Q Substitution. Clin. Appl. Thromb. Hemost. 1997, 3, 168–173. [Google Scholar] [CrossRef]
- Naess, I.A.; Christiansen, S.C.; Romundstad, P.; Cannegieter, S.C.; Rosendaal, F.R.; Hammerstrom, J. Incidence and mortality of venous thrombosis: A population-based study. J. Thromb. Haemost. 2007, 5, 692–699. [Google Scholar] [CrossRef]
- Douketis, J.; Tosetto, A.; Marcucci, M.; Baglin, T.; Cosmi, B.; Cushman, M.; Kyrle, P.; Poli, D.; Tait, R.C.; Iorio, A. Risk of recurrence after venous thromboembolism in men and women: Patient level meta-analysis. BMJ 2011, 342, d813. [Google Scholar] [CrossRef]
- Roach, R.E.; Lijfering, W.M.; Rosendaal, F.R.; Cannegieter, S.C.; le Cessie, S. Sex difference in risk of second but not of first venous thrombosis: Paradox explained. Circulation 2014, 129, 51–56. [Google Scholar] [CrossRef]
- Gemmati, D.; Varani, K.; Bramanti, B.; Piva, R.; Bonaccorsi, G.; Trentini, A.; Manfrinato, M.C.; Tisato, V.; Care, A.; Bellini, T. “Bridging the Gap” Everything that Could Have Been Avoided If We Had Applied Gender Medicine, Pharmacogenetics and Personalized Medicine in the Gender-Omics and Sex-Omics Era. Int. J. Mol. Sci. 2019, 21, 296. [Google Scholar] [CrossRef]
- Singh, A.V.; Subhashree, L.; Milani, P.; Gemmati, D.; Zamboni, P. Interplay of iron metallobiology, metalloproteinases, and FXIII, and role of their gene variants in venous leg ulcer. Int. J. Low Extrem. Wounds 2010, 9, 166–179. [Google Scholar] [CrossRef]
- Singh, A.V.; Vyas, V.; Montani, E.; Cartelli, D.; Parazzoli, D.; Oldani, A.; Zeri, G.; Orioli, E.; Gemmati, D.; Zamboni, P. Investigation of in vitro cytotoxicity of the redox state of ionic iron in neuroblastoma cells. J. Neurosci. Rural Pract. 2012, 3, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Morange, P.E.; Tregouet, D.A. Lessons from genome-wide association studies in venous thrombosis. J. Thromb. Haemost. 2011, 9 (Suppl. 1), 258–264. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Teichert, M.; Chasman, D.I.; Heit, J.A.; Morange, P.E.; Li, G.; Pankratz, N.; Leebeek, F.W.; Pare, G.; de Andrade, M.; et al. A genome-wide association study for venous thromboembolism: The extended cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Genet Epidemiol. 2013, 37, 512–521. [Google Scholar] [CrossRef] [PubMed]
- de Haan, H.G.; van Hylckama Vlieg, A.; Germain, M.; Baglin, T.P.; Deleuze, J.-F.; Trégouët, D.-A.; Rosendaal, F.R. Genome-wide association study identifies a novel genetic risk factor for recurrent venous thrombosis. Circ. Genom. Precis. Med. 2018, 11, e001827. [Google Scholar] [CrossRef]
- Zoller, B.; Svensson, P.J.; Dahlback, B.; Lind-Hallden, C.; Hallden, C.; Elf, J. Genetic risk factors for venous thromboembolism. Expert Rev. Hematol. 2020, 13, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Prandoni, P. Acquired risk factors for venous thromboembolism in medical patients. Hematol. Am. Soc. Hematol. Educ. Program. 2005. [Google Scholar] [CrossRef]
- Soria, J.M.; Morange, P.E.; Vila, J.; Souto, J.C.; Moyano, M.; Tregouet, D.A.; Mateo, J.; Saut, N.; Salas, E.; Elosua, R. Multilocus genetic risk scores for venous thromboembolism risk assessment. J. Am. Hearth Assoc. 2014, 3, e001060. [Google Scholar] [CrossRef]
- Varga, E.A.; Kujovich, J.L. Management of inherited thrombophilia: Guide for genetics professionals. Clin. Genet. 2012, 81, 7–17. [Google Scholar] [CrossRef]
- Hlatky, M.A.; Greenland, P.; Arnett, D.K.; Ballantyne, C.M.; Criqui, M.H.; Elkind, M.S.; Go, A.S.; Harrell, F.E., Jr.; Hong, Y.; Howard, B.V.; et al. Criteria for evaluation of novel markers of cardiovascular risk: A scientific statement from the American Heart Association. Circulation 2009, 119, 2408–2416. [Google Scholar] [CrossRef]
- McRae, S.; Tran, H.; Schulman, S.; Ginsberg, J.; Kearon, C. Effect of patient’s sex on risk of recurrent venous thromboembolism: A meta-analysis. Lancet 2006, 368, 371–378. [Google Scholar] [CrossRef]
- Corral, J.; de la Morena-Barrio, M.E.; Vicente, V. The genetics of antithrombin. Thromb. Res. 2018, 169, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.A.; Mannucci, P.M.; Bauer, K.A.; Bertina, R.M.; Bochkov, N.P.; Boulyjenkov, V.; Chandy, M.; Dahlback, B.; Ginter, E.K.; Miletich, J.P.; et al. Inherited thrombophilia: Part 2. Thromb. Haemost. 1996, 76, 824–834. [Google Scholar] [PubMed]
- Lane, D.A.; Mannucci, P.M.; Bauer, K.A.; Bertina, R.M.; Bochkov, N.P.; Boulyjenkov, V.; Chandy, M.; Dahlback, B.; Ginter, E.K.; Miletich, J.P.; et al. Inherited thrombophilia: Part 1. Thromb. Haemost. 1996, 76, 651–662. [Google Scholar] [PubMed]
- Bravo-Perez, C.; de la Morena-Barrio, M.E.; Palomo, A.; Entrena, L.; de la Morena-Barrio, B.; Padilla, J.; Minano, A.; Navarro, E.; Cifuentes, R.; Corral, J.; et al. Genotype-phenotype gradient of SERPINC1 variants in a single family reveals a severe compound antithrombin deficiency in a dead embryo. Br. J. Haematol. 2020, 191, e32–e35. [Google Scholar] [CrossRef]
- Chowdhury, V.; Olds, R.J.; Lane, D.A.; Conard, J.; Pabinger, I.; Ryan, K.; Bauer, K.A.; Bhavnani, M.; Abildgaard, U.; Finazzi, G.; et al. Identification of nine novel mutations in type I antithrombin deficiency by heteroduplex screening. Br. J. Haematol. 1993, 84, 656–661. [Google Scholar] [CrossRef]
- Castaldo, G.; Cerbone, A.M.; Guida, A.; Tandurella, I.; Ingino, R.; Tufano, A.; Ceglia, C.; Di Minno, M.N.; Ruocco, A.L.; Di Minno, G. Molecular analysis and genotype-phenotype correlation in patients with antithrombin deficiency from Southern Italy. Thromb. Haemost. 2012, 107, 673–680. [Google Scholar] [CrossRef]
- Duga, S.; Asselta, R.; Tenchini, M.L. Coagulation factor V. Int. J. Biochem. Cell Biol. 2004, 36, 1393–1399. [Google Scholar] [CrossRef]
- Bertina, R.M.; Koeleman, B.P.; Koster, T.; Rosendaal, F.R.; Dirven, R.J.; de Ronde, H.; van der Velden, P.A.; Reitsma, P.H. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994, 369, 64–67. [Google Scholar] [CrossRef]
- Dahlback, B.; Carlsson, M.; Svensson, P.J. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: Prediction of a cofactor to activated protein C. Proc. Natl. Acad. Sci. USA 1993, 90, 1004–1008. [Google Scholar] [CrossRef] [PubMed]
- Lunghi, B.; Iacoviello, L.; Gemmati, D.; Dilasio, M.G.; Castoldi, E.; Pinotti, M.; Castaman, G.; Redaelli, R.; Mariani, G.; Marchetti, G.; et al. Detection of new polymorphic markers in the factor V gene: Association with factor V levels in plasma. Thromb. Haemost. 1996, 75, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, F.; Faioni, E.M.; Castoldi, E.; Lunghi, B.; Castaman, G.; Sacchi, E.; Mannucci, P.M. A factor V genetic component differing from factor V R506Q contributes to the activated protein C resistance phenotype. Blood 1997, 90, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, E.; Lunghi, B.; Mingozzi, F.; Muleo, G.; Redaelli, R.; Mariani, G.; Bernardi, F. A missense mutation (Y1702C) in the coagulation factor V gene is a frequent cause of factor V deficiency in the Italian population. Haematologica 2001, 86, 629–633. [Google Scholar] [PubMed]
- Simioni, P.; Scudeller, A.; Radossi, P.; Gavasso, S.; Girolami, B.; Tormene, D.; Girolami, A. Pseudo homozygous activated protein C resistance due to double heterozygous factor V defects (factor V Leiden mutation and type I quantitative factor V defect) associated with thrombosis: Report of two cases belonging to two unrelated kindreds. Thromb. Haemost. 1996, 75, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Vos, H.L. Inherited defects of coagulation Factor V: The thrombotic side. J. Thromb. Haemost. 2006, 4, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Paraboschi, E.M.; Menegatti, M.; Rimoldi, V.; Borhany, M.; Abdelwahab, M.; Gemmati, D.; Peyvandi, F.; Duga, S.; Asselta, R. Profiling the mutational landscape of coagulation factor V deficiency. Haematologica 2020, 105, e180–e185. [Google Scholar] [CrossRef]
- Le, W.; Yu, J.D.; Lu, L.; Tao, R.; You, B.; Cai, X.; Cao, W.J.; Huang, W.; He, R.M.; Zhu, D.L.; et al. Association of the R485K polymorphism of the factor V gene with poor response to activated protein C and increased risk of coronary artery disease in the Chinese population. Clin. Genet. 2000, 57, 296–303. [Google Scholar] [CrossRef]
- Tamura, S.; Hashimoto, E.; Suzuki, N.; Kakihara, M.; Odaira, K.; Hattori, Y.; Tokoro, M.; Suzuki, S.; Takagi, A.; Katsumi, A.; et al. Molecular basis of SERPINC1 mutations in Japanese patients with antithrombin deficiency. Thromb. Res. 2019, 178, 159–170. [Google Scholar] [CrossRef]
- de Haan, H.G.; Bezemer, I.D.; Doggen, C.J.; Le Cessie, S.; Reitsma, P.H.; Arellano, A.R.; Tong, C.H.; Devlin, J.J.; Bare, L.A.; Rosendaal, F.R.; et al. Multiple SNP testing improves risk prediction of first venous thrombosis. Blood 2012, 120, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Waye, J.S.; Sheffield, W.P.; Eng, B.; Blajchman, M.A. Genetic linkage studies in antithrombin-deficient kindreds using a highly polymorphic trinucleotide short tandem repeat (STR) within the human antithrombin gene. Am. J. Hematol. 1994, 46, 107–111. [Google Scholar] [CrossRef]
- Castoldi, E.; Lunghi, B.; Mingozzi, F.; Simioni, P.; Girolami, A.; Bernardi, F. A highly polymorphic microsatellite in the factor V gene is an informative tool for the study of factor V-related disorders. Br. J. Haematol. 2001, 114, 868–870. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; Legnani, C.; Lunghi, B.; Scanavini, D.; Castoldi, E.; Palareti, G.; Marchetti, G.; Bernardi, F. A FV multiallelic marker detects genetic components of APC resistance contributing to venous thromboembolism in FV Leiden carriers. Thromb. Haemost. 2003, 89, 983–989. [Google Scholar] [PubMed]
- Quaranta, M.; Erez, O.; Mastrolia, S.A.; Koifman, A.; Leron, E.; Eshkoli, T.; Mazor, M.; Holcberg, G. The physiologic and therapeutic role of heparin in implantation and placentation. Peer. J. 2015, 3, e691. [Google Scholar] [CrossRef]
- van Boven, H.H.; Vandenbroucke, J.P.; Briet, E.; Rosendaal, F.R. Gene-gene and gene-environment interactions determine risk of thrombosis in families with inherited antithrombin deficiency. Blood 1999, 94, 2590–2594. [Google Scholar] [CrossRef] [PubMed]
- Dahm, A.E.; Bezemer, I.D.; Bergrem, A.; Jacobsen, A.F.; Jacobsen, E.M.; Skretting, G.; Rosendaal, F.R.; Sandset, P.M. Candidate gene polymorphisms and the risk for pregnancy-related venous thrombosis. Br. J. Haematol. 2012, 157, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Koster, T.; Rosendaal, F.R.; de Ronde, H.; Briet, E.; Vandenbroucke, J.P.; Bertina, R.M. Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden Thrombophilia Study. Lancet 1993, 342, 1503–1506. [Google Scholar] [CrossRef]
- Blom, J.W.; Doggen, C.J.; Osanto, S.; Rosendaal, F.R. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 2005, 293, 715–722. [Google Scholar] [CrossRef]
- Shi, X.; Xie, X.; Jia, Y.; Li, S. Maternal genetic polymorphisms and unexplained recurrent miscarriage: A systematic review and meta-analysis. Clin. Genet. 2017, 91, 265–284. [Google Scholar] [CrossRef]
- Loizidou, E.M.; Kucherenko, A.; Tatarskyy, P.; Chernushyn, S.; Livshyts, G.; Gulkovskyi, R.; Vorobiova, I.; Antipkin, Y.; Gorodna, O.; Kaakinen, M.A.; et al. Risk of Recurrent Pregnancy Loss in the Ukrainian Population Using a Combined Effect of Genetic Variants: A Case-Control Study. Genes 2021, 12, 64. [Google Scholar] [CrossRef]
- Houlihan, L.M.; Davies, G.; Tenesa, A.; Harris, S.E.; Luciano, M.; Gow, A.J.; McGhee, K.A.; Liewald, D.C.; Porteous, D.J.; Starr, J.M.; et al. Common variants of large effect in F12, KNG1, and HRG are associated with activated partial thromboplastin time. Am. J. Hum. Genet. 2010, 86, 626–631. [Google Scholar] [CrossRef]
- Li, Y.; Bezemer, I.D.; Rowland, C.M.; Tong, C.H.; Arellano, A.R.; Catanese, J.J.; Devlin, J.J.; Reitsma, P.H.; Bare, L.A.; Rosendaal, F.R. Genetic variants associated with deep vein thrombosis: The F11 locus. J. Thromb. Haemost. 2009, 7, 1802–1808. [Google Scholar] [CrossRef] [PubMed]
- Morange, P.E.; Oudot-Mellakh, T.; Cohen, W.; Germain, M.; Saut, N.; Antoni, G.; Alessi, M.C.; Bertrand, M.; Dupuy, A.M.; Letenneur, L.; et al. KNG1 Ile581Thr and susceptibility to venous thrombosis. Blood 2011, 117, 3692–3694. [Google Scholar] [CrossRef] [PubMed]
- Schmaier, A.H.; Mc Crae, K.R. The plasma kallikrein-kinin system: Its evolution from contact activation. J. Thromb. Haemost. 2007, 5, 2323–2329. [Google Scholar] [CrossRef]
- Sugi, T.; Makino, T. Factor XII, kininogen and plasma prekallikrein in abnormal pregnancies. Curr. Drug Targets 2005, 6, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.Y.; Tuite, A.; Morange, P.E.; Tregouet, D.A.; Gagnon, F. The factor XII -4C>T variant and risk of common thrombotic disorders: A HuGE review and meta-analysis of evidence from observational studies. Am. J. Epidemiol. 2011, 173, 136–144. [Google Scholar] [CrossRef]
- Wei, L.K.; Griffiths, L.R.; Kooi, C.W.; Irene, L. Meta-Analysis of Factor V, Factor VII, Factor XII, and Factor XIII-A Gene Polymorphisms and Ischemic Stroke. Medicina 2019, 55, 101. [Google Scholar] [CrossRef]
- Ibrahim-Kosta, M.; Suchon, P.; Couturaud, F.; Smadja, D.; Olaso, R.; Germain, M.; Saut, N.; Goumidi, L.; Derbois, C.; Thibord, F.; et al. Minor allele of the factor V K858R variant protects from venous thrombosis only in non-carriers of factor V Leiden mutation. Sci. Rep. 2019, 9, 3750. [Google Scholar] [CrossRef] [PubMed]
- Gorbe, E.; Nagy, B.; Varadi, V.; Kiss, E.; Mattyus, I.; Rigo, J., Jr.; Papp, Z. Mutation in the factor V gene associated with inferior vena cava thrombosis in newborns. Clin. Genet. 1999, 55, 65–66. [Google Scholar] [CrossRef]
- Kostka, H.; Siegert, G.; Schwarz, T.; Gehrisch, S.; Kuhlisch, E.; Schellong, S.; Jaross, W. Frequency of polymorphisms in the B-domain of factor V gene in APC-resistant patients. Thromb. Res. 2000, 99, 539–547. [Google Scholar] [CrossRef]
- Gemmati, D.; Tognazzo, S.; Catozzi, L.; Federici, F.; De Palma, M.; Gianesini, S.; Scapoli, G.L.; De Mattei, M.; Liboni, A.; Zamboni, P. Influence of gene polymorphisms in ulcer healing process after superficial venous surgery. J. Vasc. Surg. 2006, 44, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Tognazzo, S.; Gemmati, D.; Palazzo, A.; Catozzi, L.; Carandina, S.; Legnaro, A.; Tacconi, G.; Scapoli, G.L.; Zamboni, P. Prognostic role of factor XIII gene variants in nonhealing venous leg ulcers. J. Vasc. Surg. 2006, 44, 815–819. [Google Scholar] [CrossRef]
- Ansani, L.; Marchesini, J.; Pestelli, G.; Luisi, G.A.; Scillitani, G.; Longo, G.; Milani, D.; Serino, M.L.; Tisato, V.; Gemmati, D. F13A1 Gene Variant (V34L) and Residual Circulating FXIIIA Levels Predict Short- and Long-Term Mortality in Acute Myocardial Infarction after Coronary Angioplasty. Int. J. Mol. Sci. 2018, 19, 2766. [Google Scholar] [CrossRef] [PubMed]
- Gemmati, D.; Zeri, G.; Orioli, E.; Mari, R.; Moratelli, S.; Vigliano, M.; Marchesini, J.; Grossi, M.E.; Pecoraro, A.; Cuneo, A.; et al. Factor XIII-A dynamics in acute myocardial infarction: A novel prognostic biomarker? Thromb. Haemost. 2015, 114, 123–132. [Google Scholar] [CrossRef]
- Agostinis, C.; Bulla, R.; Tisato, V.; De Seta, F.; Alberico, S.; Secchiero, P.; Zauli, G. Soluble TRAIL is elevated in recurrent miscarriage and inhibits the in vitro adhesion and migration of HTR8 trophoblastic cells. Hum. Reprod. 2012, 27, 2941–2947. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
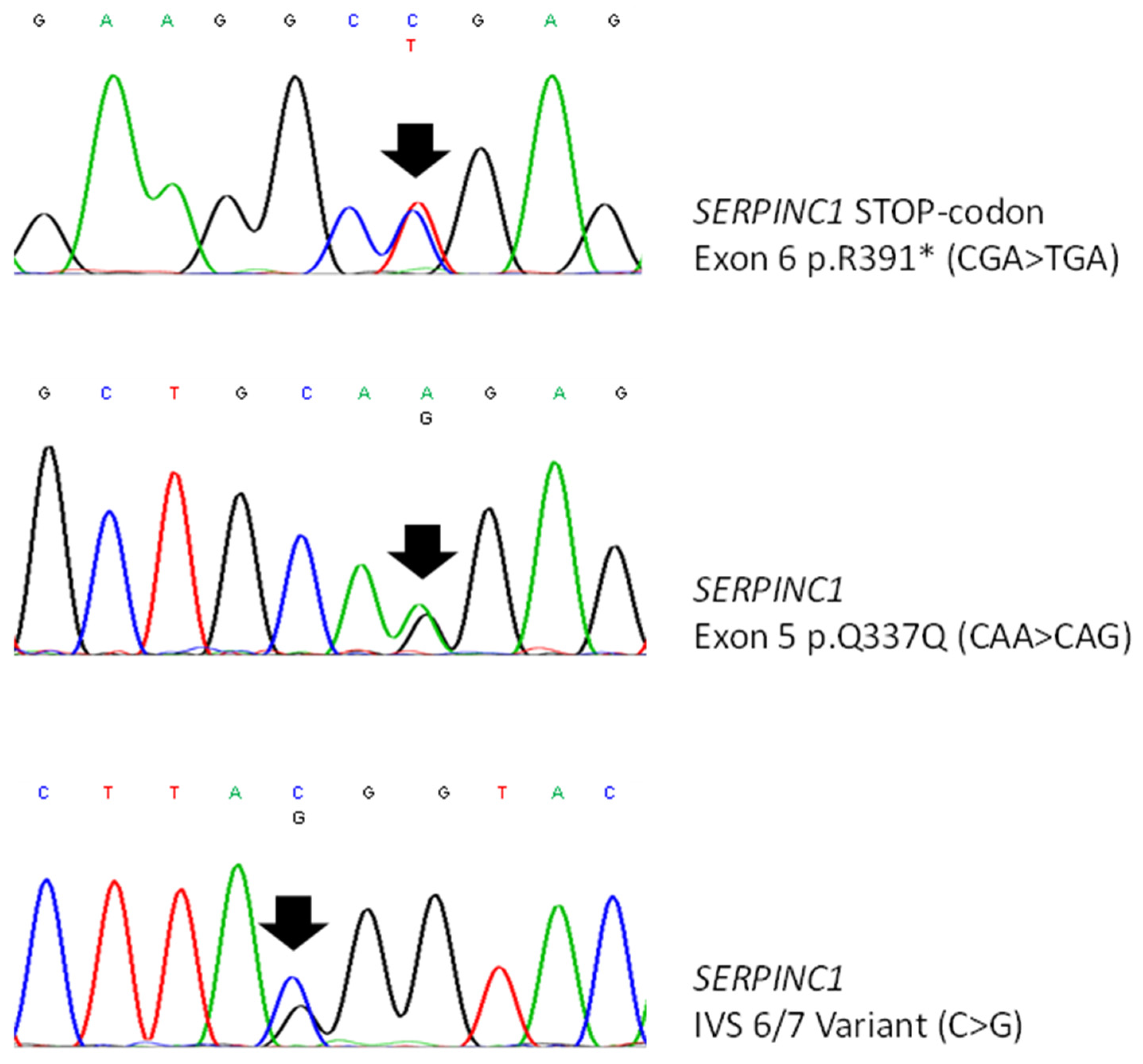{kind=link}
| Ch. | Gene | Change (nt) | MAF | Variant | Change (aa) | Family 1b | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| II2 | II3 | III1 | III2 | III3 | IV1 | IV2 | ||||||
| 9 | ABO rs8176719 | −/G | G (0.379) | c.261delG | − | −/− | −/− | −/− | −/− | −/− | −/− | −/− |
| 11 | F2 rs1799963 | G > A | A (0.0135) | 3′ UTR | − | GG | GG | GG | GG | GG | GG | GG |
| 1 | F5 rs6025 | G > A | A (0.025) | Missense | R506Q | GA | GG | GA | GA | GA | GA | GA |
| 1 | F5 rs1800595 | A > G | G (0.0485) | Missense | H1299R | AA | AA | AA | AA | AA | AA | AG |
| 1 | F5 rs4524 | T > C | C (0.267) | Missense | K830R | TC | TT | TT | TT | TT | TT | TC |
| 4 | F11 rs2289252 | G > A | A (0.399) | Intron | − | AA | AA | AG | AA | AA | AA | AA |
| 4 | F11 rs2036914 | G > A | A (0.479) | Intron | − | GG | GG | GA | GG | GG | GG | GG |
| 5 | F12 rs1801020 | G > A | A (0.237) | 5′ UTR | − | GG | GA | GA | GA | GA | GA | GA |
| 6 | F13A1 rs5985 | G > T | T (0.243) | Missense | V34L | GG | GT | GG | GT | GT | GT | GT |
| 1 | SERPINE10 rs2232698 | G > A | A (0.0077) | Stop Gained | R67 * | GG | GG | GG | GG | GG | GG | GG |
| 1 | SERPINC1 rs121909548 | G > T | T (0.0015) | Missense | A384S | GG | GG | GG | GG | GG | GG | GG |
| 4 | FGG rs2066865 | C > T | T (0.23) | near 3′UTR | − | CC | CC | CC | CC | CC | CC | CC |
| 3 | KNG1 rs710446 | T > C | C (0.419) | Missense | I581T | TC | TC | TT | CC | TC | TC | TC |
| Gene | Variation | Family 1b | ||||||
|---|---|---|---|---|---|---|---|---|
| II2 | II3 | III1 | III2 | III3 | IV1 | IV2 | ||
| SERPINC1 | c.1171C>T (p.R391*) | CT | CC | CC | CT | CT | CT | CT |
| SERPINC1 | c.1011A>G (p.Q337Q) rs5878 | AA | AG | AG | AG | AG | AG | AG |
| SERPINC1 | (rs677) C > G | CG | CG | GG | CG | CG | CG | CG |
| SERPINC1 | IVS5 (ATT)5–18 | 10/12 | 11/13 | 10/11 | 11/12 | 11/12 | 10/12 | nd |
| F5 | c.1691A>G (p.R506Q) | AG | AA | AG | AG | AG | AG | AG |
| F5 | IVS2 (AT)6–33 | 15/17 | 15/19 | 16/20 | 17/19 | 17/19 | 16/19 | nd |
| F5 | IVS11 (GT)12–16 | 14/15 | 13/15 | 13/14 | 13/14 | 13/14 | 13/14 | 14/14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gemmati, D.; Longo, G.; Franchini, E.; Araujo Silva, J.; Gallo, I.; Lunghi, B.; Moratelli, S.; Maestri, I.; Serino, M.L.; Tisato, V. Cis-Segregation of c.1171C>T Stop Codon (p.R391*) in SERPINC1 Gene and c.1691G>A Transition (p.R506Q) in F5 Gene and Selected GWAS Multilocus Approach in Inherited Thrombophilia. Genes 2021, 12, 934. https://doi.org/10.3390/genes12060934
Gemmati D, Longo G, Franchini E, Araujo Silva J, Gallo I, Lunghi B, Moratelli S, Maestri I, Serino ML, Tisato V. Cis-Segregation of c.1171C>T Stop Codon (p.R391*) in SERPINC1 Gene and c.1691G>A Transition (p.R506Q) in F5 Gene and Selected GWAS Multilocus Approach in Inherited Thrombophilia. Genes. 2021; 12(6):934. https://doi.org/10.3390/genes12060934
Chicago/Turabian StyleGemmati, Donato, Giovanna Longo, Eugenia Franchini, Juliana Araujo Silva, Ines Gallo, Barbara Lunghi, Stefano Moratelli, Iva Maestri, Maria Luisa Serino, and Veronica Tisato. 2021. "Cis-Segregation of c.1171C>T Stop Codon (p.R391*) in SERPINC1 Gene and c.1691G>A Transition (p.R506Q) in F5 Gene and Selected GWAS Multilocus Approach in Inherited Thrombophilia" Genes 12, no. 6: 934. https://doi.org/10.3390/genes12060934
APA StyleGemmati, D., Longo, G., Franchini, E., Araujo Silva, J., Gallo, I., Lunghi, B., Moratelli, S., Maestri, I., Serino, M. L., & Tisato, V. (2021). Cis-Segregation of c.1171C>T Stop Codon (p.R391*) in SERPINC1 Gene and c.1691G>A Transition (p.R506Q) in F5 Gene and Selected GWAS Multilocus Approach in Inherited Thrombophilia. Genes, 12(6), 934. https://doi.org/10.3390/genes12060934