
Variants in MHY7 Gene Cause Arrhythmogenic Cardiomyopathy

 ,
, 
 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.1.1. Family A
2.1.2. Family B
2.2. Next-Generation Sequencing (NGS)
2.3. In Silico Analysis
3. Results
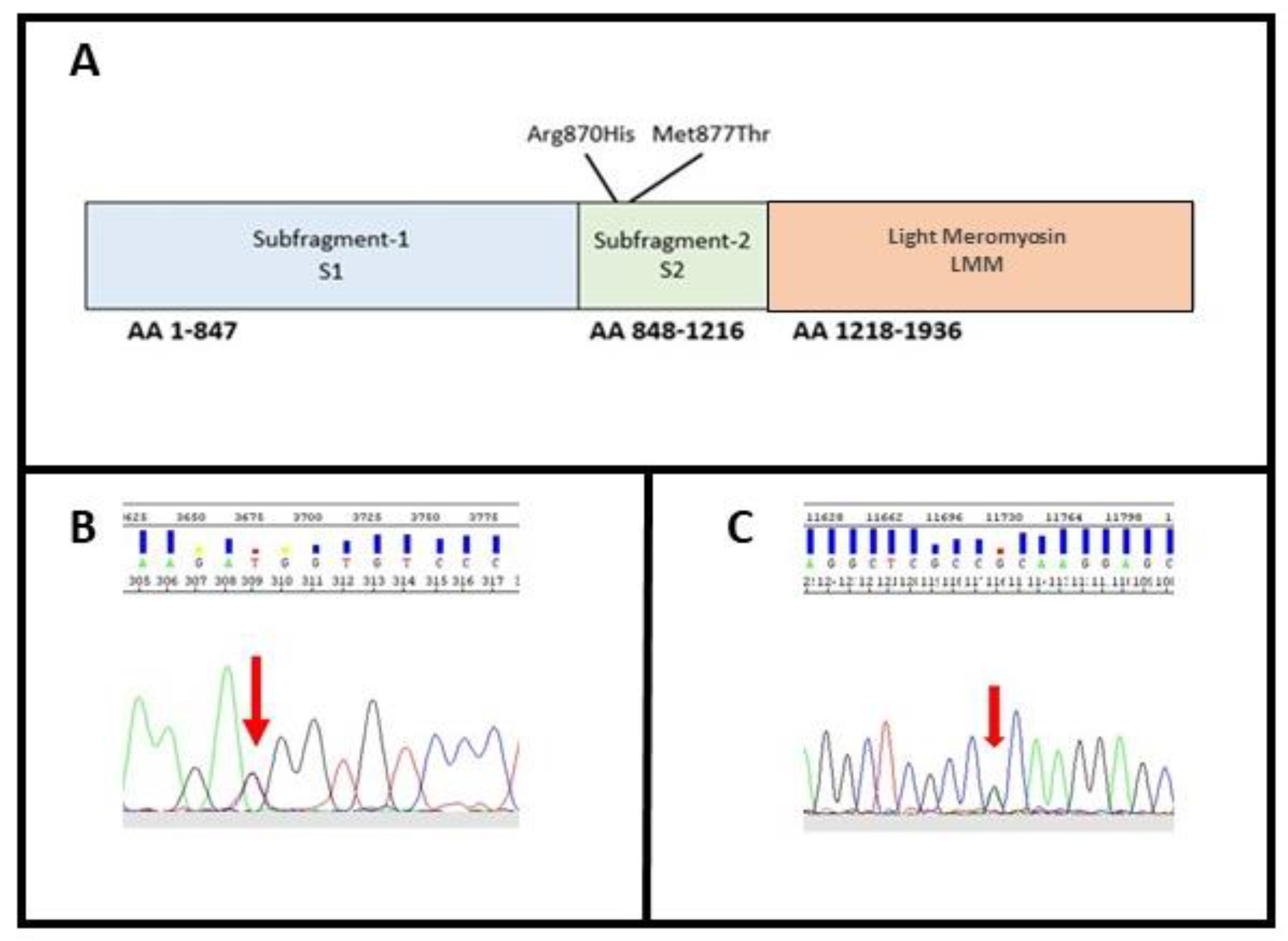
3.1. Molecular Characterization
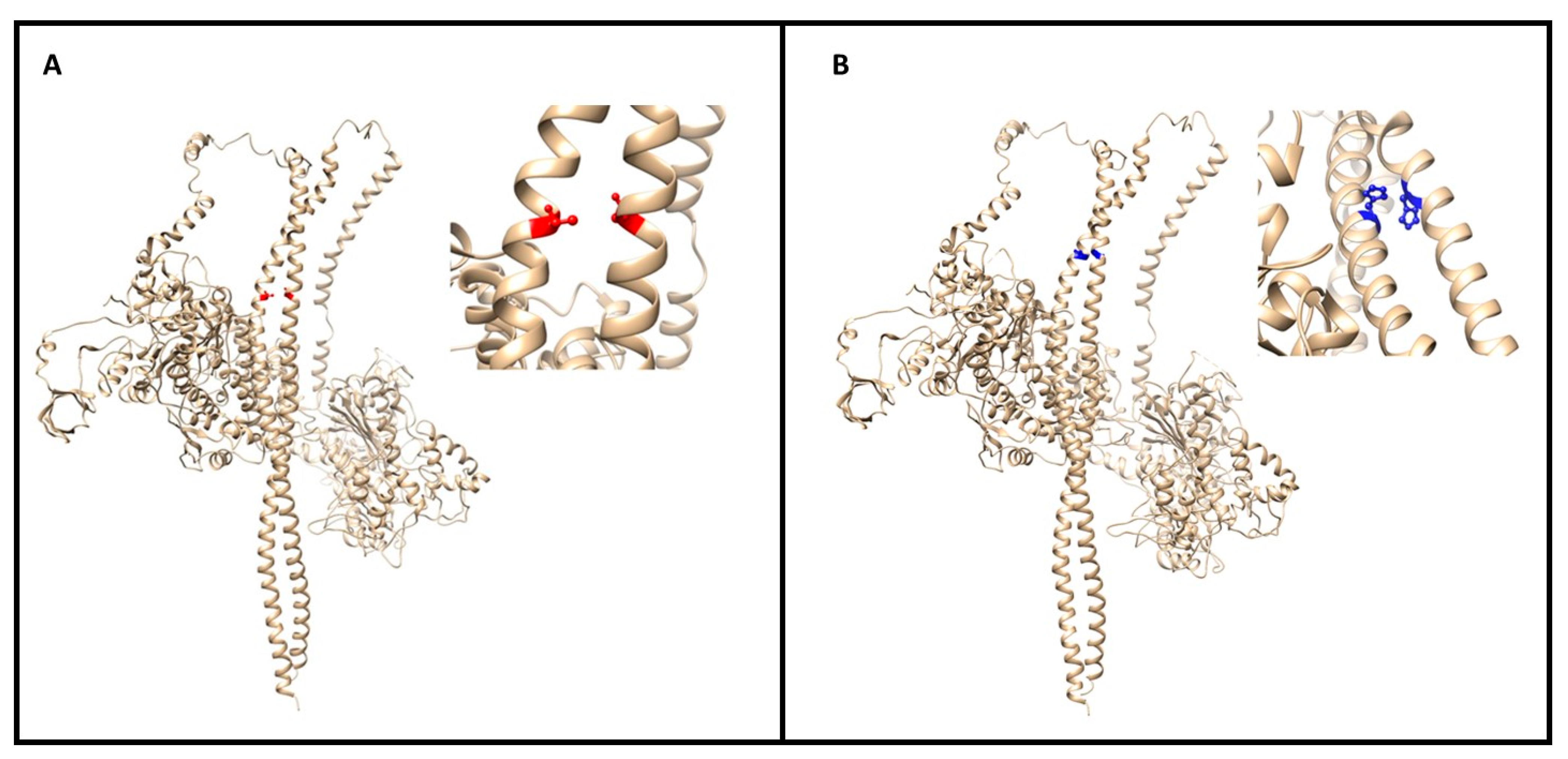
3.2. In Silico Characterization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right Ventricular Cardiomyopathy and Sudden Death in Young People. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Bauce, B.; Corrado, D.; Thiene, G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2012, 9, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; Ward, D.; Asimaki, A.; Sevdalis, E.; McKenna, W.J. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 2007, 115, 1710–1720. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Hoorntje, E.T.; Rijdt, W.P.T.; James, C.A.; Pilichou, K.; Basso, C.; Judge, D.P.; Bezzina, C.R.; Van Tintelen, J.P. Arrhythmogenic cardiomyopathy: Pathology, genetics, and concepts in pathogenesis. Cardiovasc. Res. 2017, 113, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef]
- Bhonsale, A.; Groeneweg, J.A.; James, C.A.; Dooijes, D.; Tichnell, C.; Jongbloed, J.D.H.; Murray, B.; Riele, A.S.J.M.T.; Berg, M.P.V.D.; Bikker, H.; et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Hear. J. 2015, 36, 847–855. [Google Scholar] [CrossRef]
- Nava, A.; Bauce, B.; Basso, C.; Muriago, M.; Rampazzo, A.; Villanova, C.; Daliento, L.; Buja, G.; Corrado, D.; Danieli, G.A.; et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2000, 36, 2226–2233. [Google Scholar] [CrossRef]
- Poloni, G.; De Bortoli, M.; Calore, M.; Rampazzo, A.; Lorenzon, A. Arrhythmogenic right-ventricular cardiomyopathy: Molecular genetics into clinical practice in the era of next generation sequencing. J. Cardiovasc. Med. (Hagerstown) 2016, 17, 399–407. [Google Scholar] [CrossRef]
- Bs, K.L.T.; Bs, D.J.T.; Bos, J.M.; Haugaa, K.H.; Ackerman, M.J. Whole exome sequencing with genomic triangulation implicatesCDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit. Hear. Dis. 2017, 12, 226–235. [Google Scholar] [CrossRef]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.-C.; Chin, A.; Laing, N.; Ntusi, N.B.; et al. Identification of Cadherin 2 ( CDH2 ) Mutations in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Asatryan, B.; Siripanthong, B.; Munroe, P.B.; Tiku-Owens, A.; Lopes, L.R.; Khanji, M.Y.; Protonotarios, A.; Santangeli, P.; Muser, D.; et al. State of the Art Review on Genetics and Precision Medicine in Arrhythmogenic Cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 6615. [Google Scholar] [CrossRef] [PubMed]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in Human Desmoplakin Domain Binding to Plakoglobin Causes a Dominant Form of Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; A McDermott, D.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; A Macrae, C.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef]
- Pilichou, K.; Nava, A.; Basso, C.; Beffagna, G.; Bauce, B.; Lorenzon, A.; Frigo, G.; Vettori, A.; Valente, M.; Towbin, J.; et al. Mutations in Desmoglein-2 Gene Are Associated With Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2006, 113, 1171–1179. [Google Scholar] [CrossRef]
- Asimaki, A.; Syrris, P.; Wichter, T.; Matthias, P.; Saffitz, J.E.; McKenna, W.J. A Novel Dominant Mutation in Plakoglobin Causes Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 964–973. [Google Scholar] [CrossRef]
- Syrris, P.; Ward, D.; Evans, A.; Asimaki, A.; Gandjbakhch, E.; Sen-Chowdhry, S.; McKenna, W.J. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Associated with Mutations in the Desmosomal Gene Desmocollin-2. Am. J. Hum. Genet. 2006, 79, 978–984. [Google Scholar] [CrossRef]
- Pilichou, K.; Thiene, G.; Bauce, B.; Rigato, I.; Lazzarini, E.; Migliore, F.; Marra, M.P.; Rizzo, S.; Zorzi, A.; Daliento, L.; et al. Arrhythmogenic cardiomyopathy. Orphanet J. Rare Dis. 2016, 11, 1–17. [Google Scholar] [CrossRef]
- Medeiros-Domingo, A.; Saguner, A.M.; Magyar, I.; Bahr, A.; Akdis, D.; Brunckhorst, C.; Duru, F.; Berger, W. Arrhythmogenic right ventricular cardiomyopathy: Implications of next-generation sequencing in appropriate diagnosis. Ep Europace 2017, 19, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Forleo, C.; D’Erchia, A.M.; Sorrentino, S.; Manzari, C.; Chiara, M.; Iacoviello, M.; Guaricci, A.I.; De Santis, D.; Musci, R.L.; La Spada, A.; et al. Targeted next-generation sequencing detects novel gene–phenotype associations and expands the mutational spectrum in cardiomyopathies. PLoS ONE 2017, 12, e0181842. [Google Scholar] [CrossRef]
- Mango, R.; Luchetti, A.; Sangiuolo, R.; Ferradini, V.; Briglia, N.; Giardina, E.; Ferre’, F.; Citterich, M.H.; Romeo, F.; Novelli, G.; et al. Next Generation Sequencing and Linkage Analysis for the Molecular Diagnosis of a Novel Overlapping Syndrome Characterized by Hypertrophic Cardiomyopathy and Typical Electrical Instability of Brugada Syndrome. Circ. J. 2016, 80, 938–949. [Google Scholar] [CrossRef] [PubMed]
- De Bortoli, M.; Calore, C.; Lorenzon, A.; Calore, M.; Poloni, G.; Mazzotti, E.; Rigato, I.; Marra, M.P.; Melacini, P.; Iliceto, S.; et al. Co-inheritance of mutations associated with arrhythmogenic cardiomyopathy and hypertrophic cardiomyopathy. Eur. J. Hum. Genet. 2017, 25, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Ms, B.M.; Hoorntje, E.T.; Riele, A.S.J.M.T.; Tichnell, C.; Van Der Heijden, J.F.; Tandri, H.; Berg, M.P.V.D.; Jongbloed, J.D.H.; Wilde, A.A.M.; Hauer, R.N.W.; et al. Identification of sarcomeric variants in probands with a clinical diagnosis of arrhythmogenic right ventricular cardiomyopathy (ARVC). J. Cardiovasc. Electrophysiol. 2018, 29, 1004–1009. [Google Scholar] [CrossRef]
- Ferradini, V.; Cosma, J.; Romeo, F.; De Masi, C.; Murdocca, M.; Spitalieri, P.; Mannucci, S.; Parlapiano, G.; Di Lorenzo, F.; Martino, A.; et al. LMNA variations in Dilated Cardiomiopathy patients: genotype-phenotype correlation and functional analyses. Front. Cardiov. Med. 2021. submitted. [Google Scholar]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Briefings Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Mosca, R.; Tenorio-Laranga, J.; Olivella, R.; Alcalde, V.; Céol, A.; Soler-López, M.; Aloy, P. dSysMap: Exploring the edgetic role of disease mutations. Nat. Methods 2015, 12, 167–168. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Rousseau, F.; Martins, I.C.; Ferkinghoff-Borg, J.; Stricher, F.; Serrano, L. Prediction of water and metal binding sites and their affinities by using the Fold-X force field. Proc. Natl. Acad. Sci. 2005, 102, 10147–10152. [Google Scholar] [CrossRef]
- Palmer, B.M. Thick Filament Proteins and Performance in Human Heart Failure. Hear. Fail. Rev. 2005, 10, 187–197. [Google Scholar] [CrossRef]
- Melacini, P.; Corbetti, F.; Calore, C.; Pescatore, V.; Smaniotto, G.; Pavei, A.; Bobbo, F.; Cacciavillani, L.; Iliceto, S. Cardiovascular magnetic resonance signs of ischemia in hypertrophic cardiomyopathy. Int. J. Cardiol. 2008, 128, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Kimura, A.; Harada, H.; Koga, Y.; Adachi, K.; Matsuyama, K.; Koyanagi, T.; Yasunaga, S.; Imaizumi, T.; Toshima, H. A myosin missense mutation, not a null allele, causes familial hypertrophic cardiomyopathy. Circulation 1995, 91, 2911–2915. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J.; Asatryan, B.; Wehrens, X.H.T. Genetic basis and molecular biology of cardiac arrhythmias in cardiomyopathies. Cardiovasc. Res. 2020, 116, 1600–1619. [Google Scholar] [CrossRef]
- Yotti, R.; Seidman, C.E.; Seidman, J.G. Advances in the Genetic Basis and Pathogenesis of Sarcomere Cardiomyopathies. Annu. Rev. Genom. Hum. Genet. 2019, 20, 129–153. [Google Scholar] [CrossRef] [PubMed]
- Colegrave, M.; Peckham, M. Structural implications of beta-cardiac myosin heavy chain mutations in human disease. Anat. Rec. (Hoboken) 2014, 297, 1670–1680. [Google Scholar] [CrossRef]
- Wang, J.; Wan, K.; Sun, J.; Li, W.; Liu, H.; Han, Y.; Chen, Y. Phenotypic diversity identified by cardiac magnetic resonance in a large hypertrophic cardiomyopathy family with a single MYH7 mutation. Sci. Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Miura, F.; Shimada, J.; Kitagawa, Y.; Otani, K.; Sato, T.; Toki, T.; Takahashi, T.; Yonesaka, S.; Mizukami, H.; EtsuroIt. MYH7 mutation identified by next-generation sequencing in three infant siblings with bi-ventricular noncompaction presenting with restrictive hemodynamics: A report of three siblings with a severe phenotype and poor prognosis. J. Cardiol. Cases 2019, 19, 140–143. [Google Scholar] [CrossRef]
- Alessi, C.E.; Wu, Q.; Whitaker, C.H.; Felice, K.J. Laing Myopathy: Report of 4 New Families With Novel MYH7 Mutations, Double Mutations, and Severe Phenotype. J. Clin. Neuromuscul. Dis. 2020, 22, 22–34. [Google Scholar] [CrossRef]
- Blankenfeldt, W.; Thoma, N.H.; Wray, J.S.; Gautel, M.; Schlichting, I. Crystal structures of human cardiac beta-myosin II S2-delta provide insight into the functional role of the S2 subfragment. Proc. Natl. Acad. Sci. USA 2006, 103, 17713–17717. [Google Scholar] [CrossRef]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011, 8, 1308–1339. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fam | Sex | Age (y) | Family History | Symptoms | ECG | Contrast-Enhanced CMR | Further Examinations | Medical History and FU |
|---|---|---|---|---|---|---|---|---|
| 1 | F | 38 | Father: scd when 60 years with autoptic diagnosis of AC. Paternal grandfather: SCD when 55 years Two brothers of the paternal grandfather: SCD at 49 and 64 years respectively. Sister: biventricular AC; AICD recipient | Frequent and complex PVB during exercise. NYHA I class. | Diffuse low QRS voltages.Epsilon wave in V1-V3 leads. TWI in 2-V6 leads. | RV dysfunction (RVEF 35%). LV dysfunction (LVEF 41%); apical hypokinesis; infero-distal wall akinesis. Subepicardial mid-basal anterior LV wall LGE; subepi-intramyocardial infero-lateral LV wall LGE; transmural LV apex LGE | Normal coronary computed tomography. ECG Holter (2019): 4693 polymorphic PVBs (prevalent origin from the RVOT9; 78 ventricular couplets | Subcutaneous AICD implanted for primary prevention (2014). No SVT in treatment with sotalol during 6 y of FU. |
| 1 | F | 37 | Father: scd at 60 years with autoptic diagnosis of AC. Paternal grandfather: SCD at 55 years. Two brothers of the paternal grandfather: SCD at 49 and 64 years respectively. Sister: biventricular AC; AICD recipient | Palpitations. NYHA I class. | Low QRS voltages in DI and avL leads | Not done | TTE (2019): normal LVEF (65%); normal RVd2 diameter (23 mm): mild mitral, tricuspidal and pulmonary regurgitation | No syncopal or major arrhythmic episodes during 2 y of FU. |
| 2 | M | 52 | Negative for SCD/AC | Syncopal episodes and SVT (2005).NYHA II class. | Diffuse low QRS voltagesEpsilon wave in V1-V3 leads TWI in V2-V6 leads. | RV dysfunction (RVEF 35%). LV dysfunction (LVEF 41%); apical hypokinesis; infero-distal wall akinesis. Subepicardial mid-basal anterior LV wall LGE; subepi-intramyocardial infero-lateral LV wall LGE; transmural apex LGE | TTE (2007): normal LVEF (60%); enlarged RV; mitral valve prolapsed with mild regurgitation. Coronary angiography: normal (2005). Positive EPS (2007). | Dual-chamber AICD implanted (2007). Previous appropriate AICD shock due to SVT (2007). No further SVT/FV during 13 y of FU |
| 2 | F | 18 | Father: biventricular AC, AICD recipient | Holt-Oram syndrome;Type II Mellitus diabetes; Syncopal episodes; Palpitations;NYHA I class | Normal | CMR (2019): normal; no LGE | TTE (2019): normal. Holter ECG (2020): no ventricular arrhythmias | ILR implantations; episodes of sinus tachycardia during palpitations and during syncopal episodes (1 y of FU) |
| Variant | c.2630T>C | c.2609G>A |
|---|---|---|
| ACMG classification | Likely pathogenic | Pathogenic |
| DANN | 0.9858 | 0.9996 |
| MutationTaster | Disease causing | Disease causing |
| FATHMM | Damaging | Damaging |
| MetaSVM | Damaging | Damaging |
| GERP | 4.73 | 4.73 |
| SIFT | Damaging | Damaging |
| Provean | Damaging | Damaging |
| Frequency | / | 0.00000398 |
| Species | Match | AA | Alignment |
|---|---|---|---|
| Human | 877 | SEARRKELEEKMVSLLQEKNDLQL | |
| Human mutated | not conserved | 877 | SEARRKELEEKTVSLLQEKNDLQ |
| P. troglodytes | no homologue | ||
| M. mulatta | all identical | 869 | SEARRKELEEKMVSLLQEKNDLQ |
| F. catus | all identical | 810 | SEARRKELEEKMVSLLQEKNDLQ |
| M. musculus | all identical | 877 | SEARRKELEEKMVSLLQEKNDLQ |
| G. gallus | all identical | 883 | SEARRKELEEKMVSLLQEKNDLQ |
| T. rubripes | no homologue | ||
| D. rerio | all identical | 879 | SEARRKELEEKMVSLLQEKNDLQ |
| D. melanogaster | no homologue | ||
| C. elegans | no homologue | ||
| X. tropicalis | all identical | 877 | SEARRKELEEKMVSLLQEKNDLQ |
| Species | Match | AA | Alignment |
|---|---|---|---|
| Human | 870 | LKEALEKSEARRKELEEKMVSLLQ | |
| Human mutated | not conserved | 870 | LKEALEKSEARHKELEEKMVSLL |
| P. troglodytes | no homologue | ||
| M. mulatta | all identical | 862 | LKEALEKSEARRKELEEKMVSLL |
| F. catus | all identical | 803 | LKEALEKSEARRKELEEKMVSLL |
| M. musculus | all identical | 870 | LKEALEKSEARRKELEEKMVSLL |
| G. gallus | all identical | 876 | LKEALEKSEARRKELEEKMVSLL |
| T. rubripes | no homologue | ||
| D. rerio | all identical | 872 | LKEALEKSEARRKELEEKMVSLL |
| D. melanogaster | no homologue | ||
| C. elegans | no homologue | ||
| X. tropicalis | all identical | 870 | LKEALEKSEARRKELEEKMVSLL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferradini, V.; Parca, L.; Martino, A.; Lanzillo, C.; Silvetti, E.; Calò, L.; Caselli, S.; Novelli, G.; Helmer-Citterich, M.; Sangiuolo, F.C.; et al. Variants in MHY7 Gene Cause Arrhythmogenic Cardiomyopathy. Genes 2021, 12, 793. https://doi.org/10.3390/genes12060793
Ferradini V, Parca L, Martino A, Lanzillo C, Silvetti E, Calò L, Caselli S, Novelli G, Helmer-Citterich M, Sangiuolo FC, et al. Variants in MHY7 Gene Cause Arrhythmogenic Cardiomyopathy. Genes. 2021; 12(6):793. https://doi.org/10.3390/genes12060793
Chicago/Turabian StyleFerradini, Valentina, Luca Parca, Annamaria Martino, Chiara Lanzillo, Elisa Silvetti, Leonardo Calò, Stefano Caselli, Giuseppe Novelli, Manuela Helmer-Citterich, Federica Carla Sangiuolo, and et al. 2021. "Variants in MHY7 Gene Cause Arrhythmogenic Cardiomyopathy" Genes 12, no. 6: 793. https://doi.org/10.3390/genes12060793
APA StyleFerradini, V., Parca, L., Martino, A., Lanzillo, C., Silvetti, E., Calò, L., Caselli, S., Novelli, G., Helmer-Citterich, M., Sangiuolo, F. C., & Mango, R. (2021). Variants in MHY7 Gene Cause Arrhythmogenic Cardiomyopathy. Genes, 12(6), 793. https://doi.org/10.3390/genes12060793