
Pilot Screening of Cell-Free mtDNA in NIPT: Quality Control, Variant Calling, and Haplogroup Determination

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Data
2.2. Plasma DNA Isolation
2.3. Buffy Coat Analysis
2.4. Library Preparation & Sequencing
2.5. Variant Calling and ClinVar Annotation
2.6. Haplogroup Assignment
3. Results
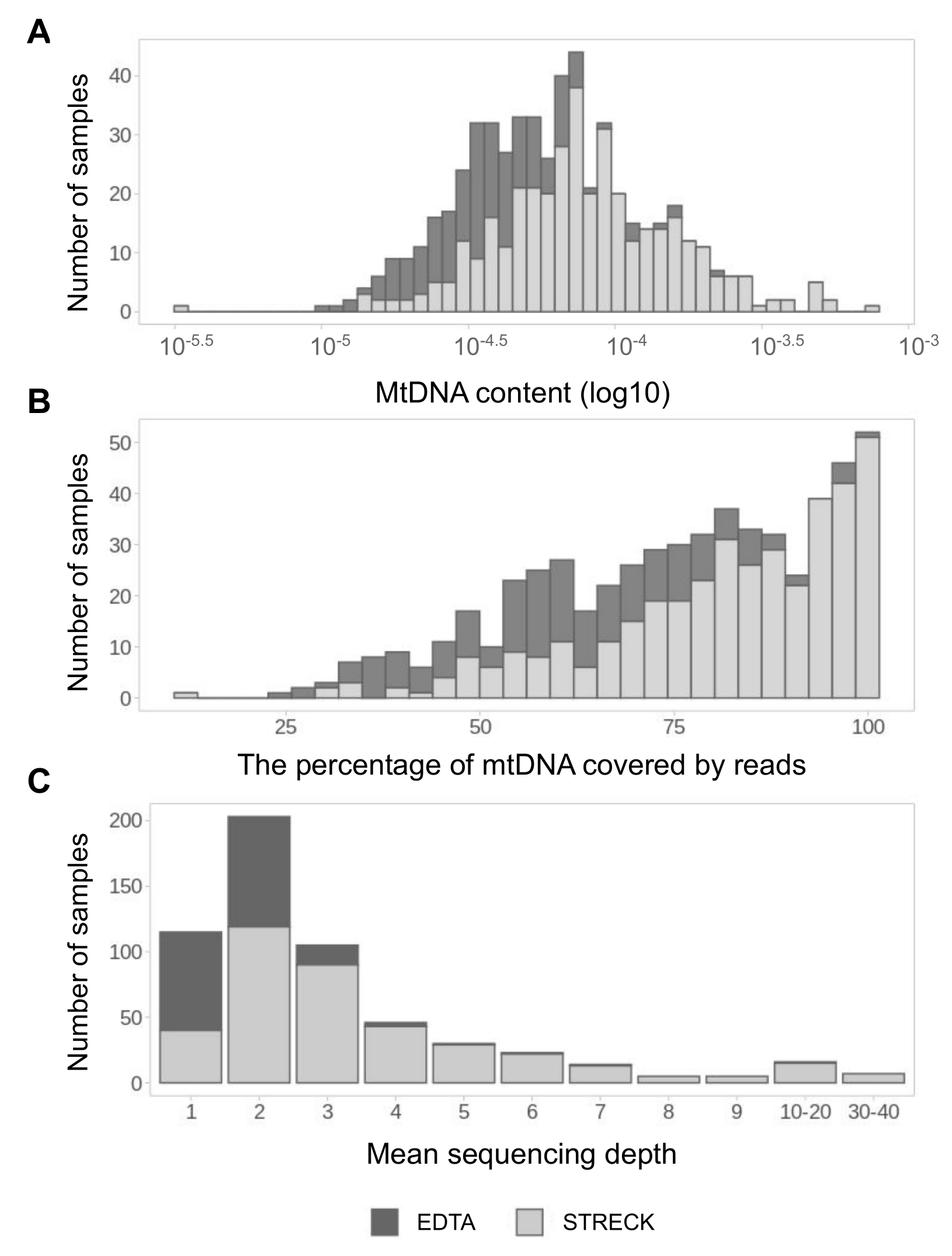
3.1. MtDNA Pool Is Better Presented in cfDNA Samples from Streck Collection Tubes
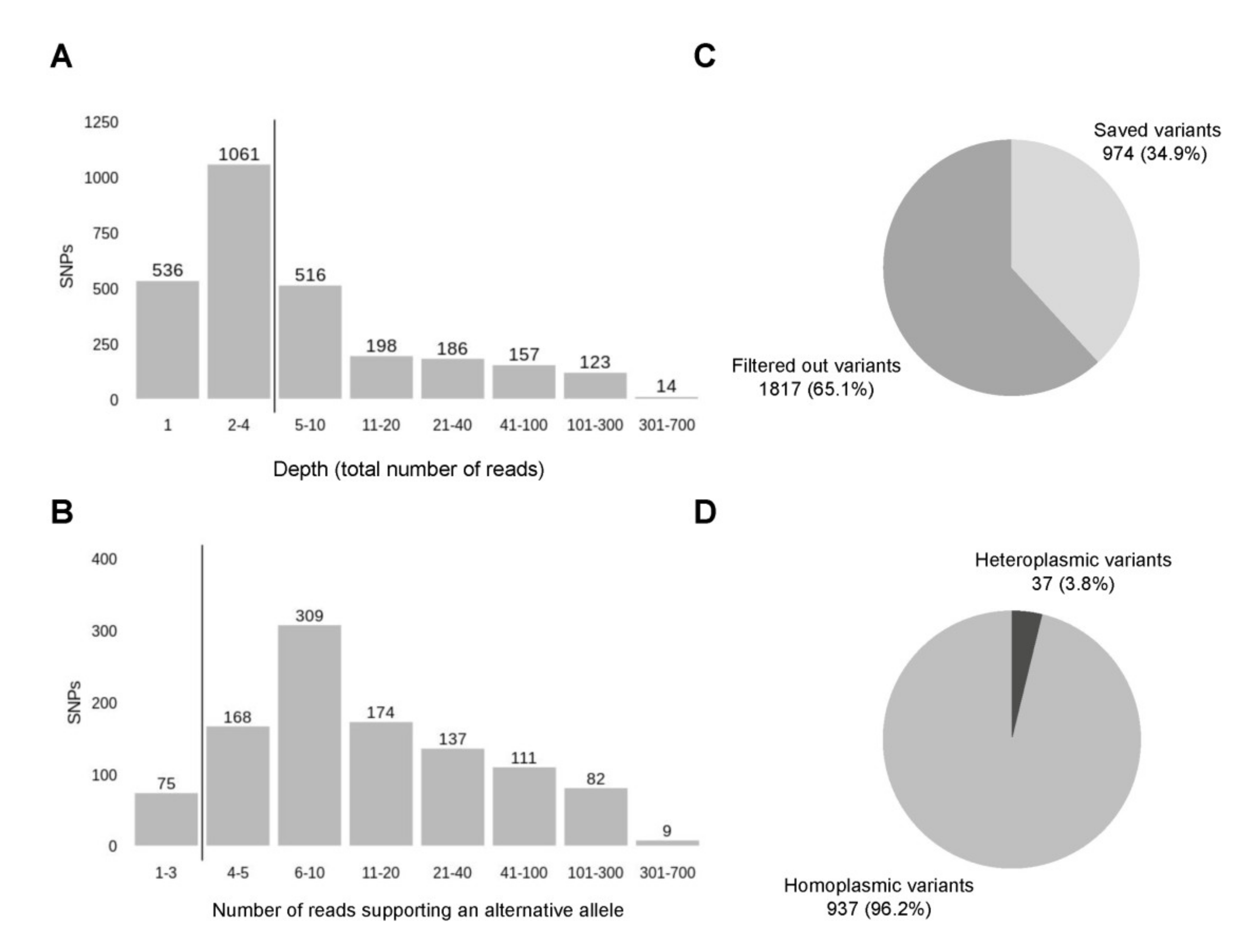
3.2. Variant Calling Reveals a Number of Non-Ancestral Mitochondrial Variants Frequent within the Russian Population
3.3. The Buffy Coat and cfDNA Contain Different Pools of mtDNA
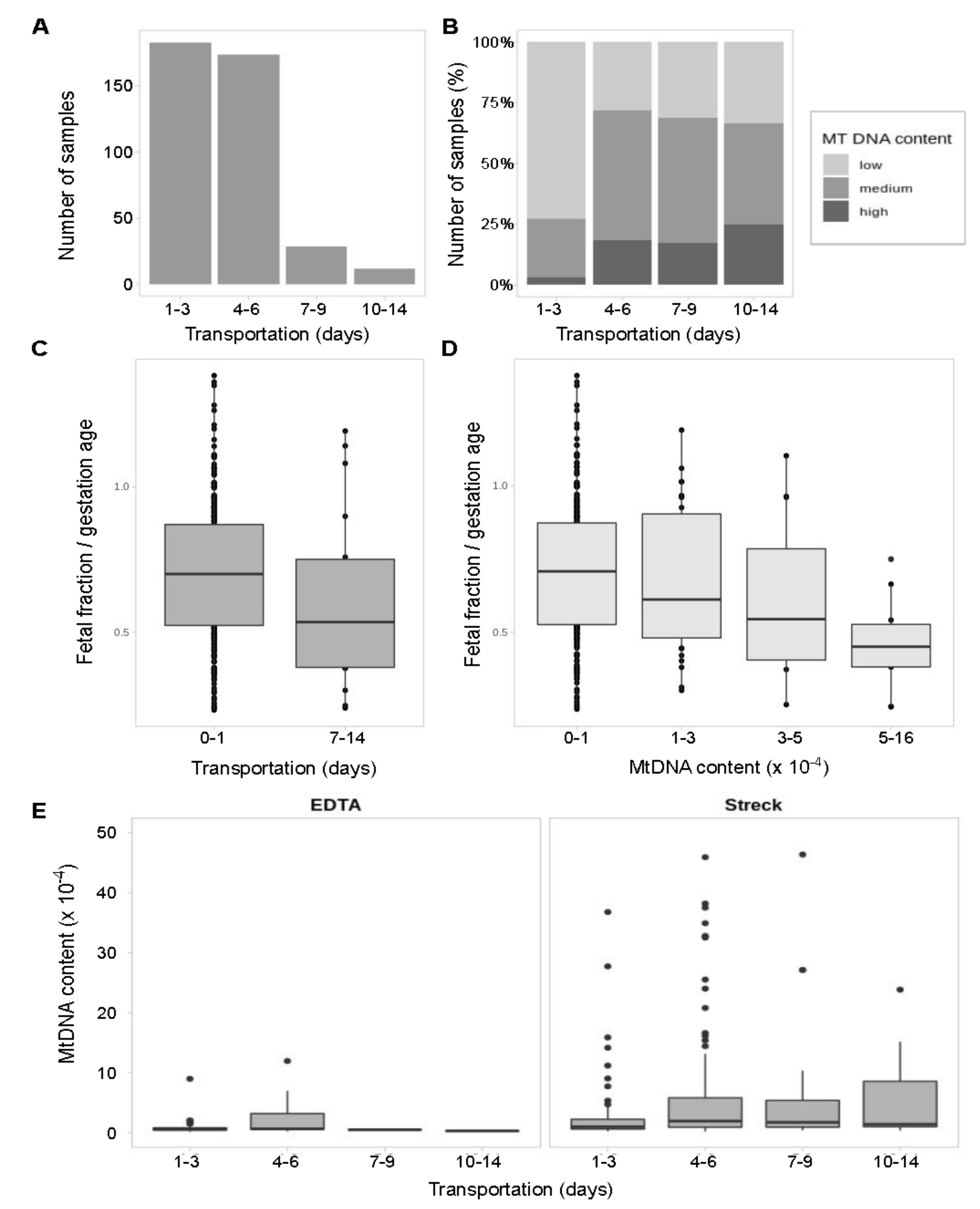
3.4. Long Transportation Results in an Increase of mtDNA Content which Leads to the Lower Fetal Fraction in Samples
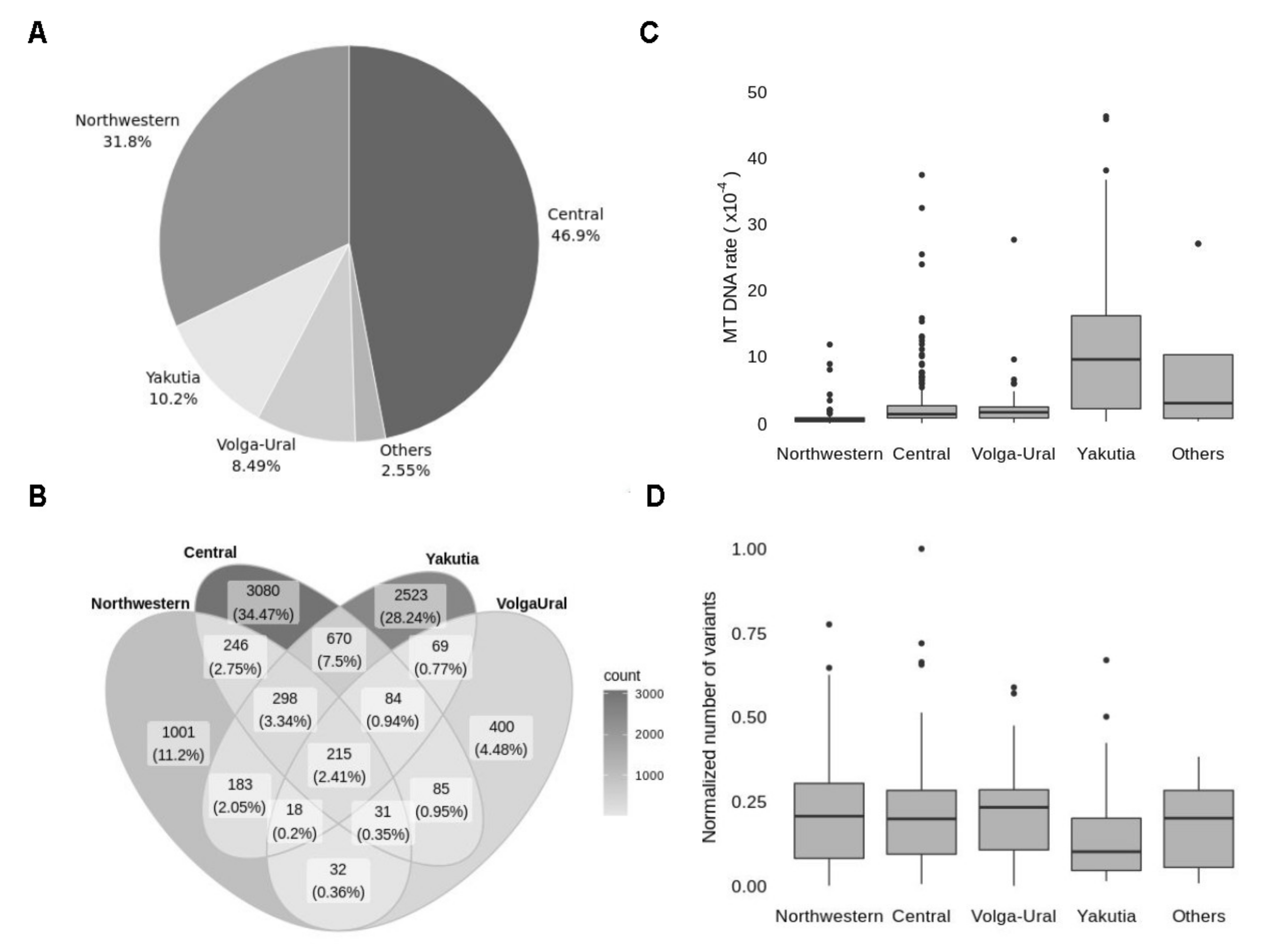
3.5. Analysis of Population Frequencies Reveals Differences in Top-Frequent Variants between Regions of Russia
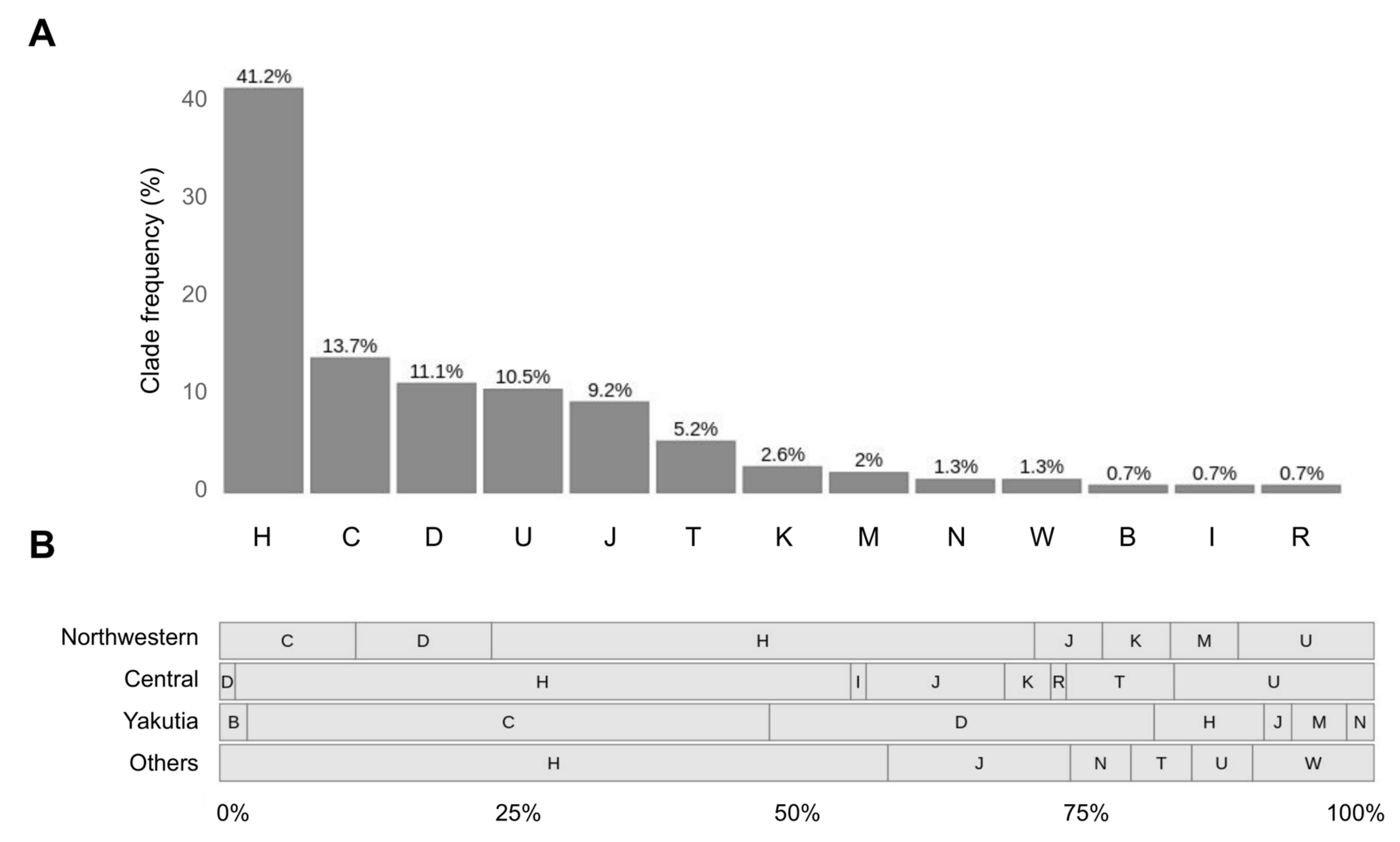
3.6. MtDNA Haplogroup H along with C, D and U Are the Most Frequent among Explored Samples
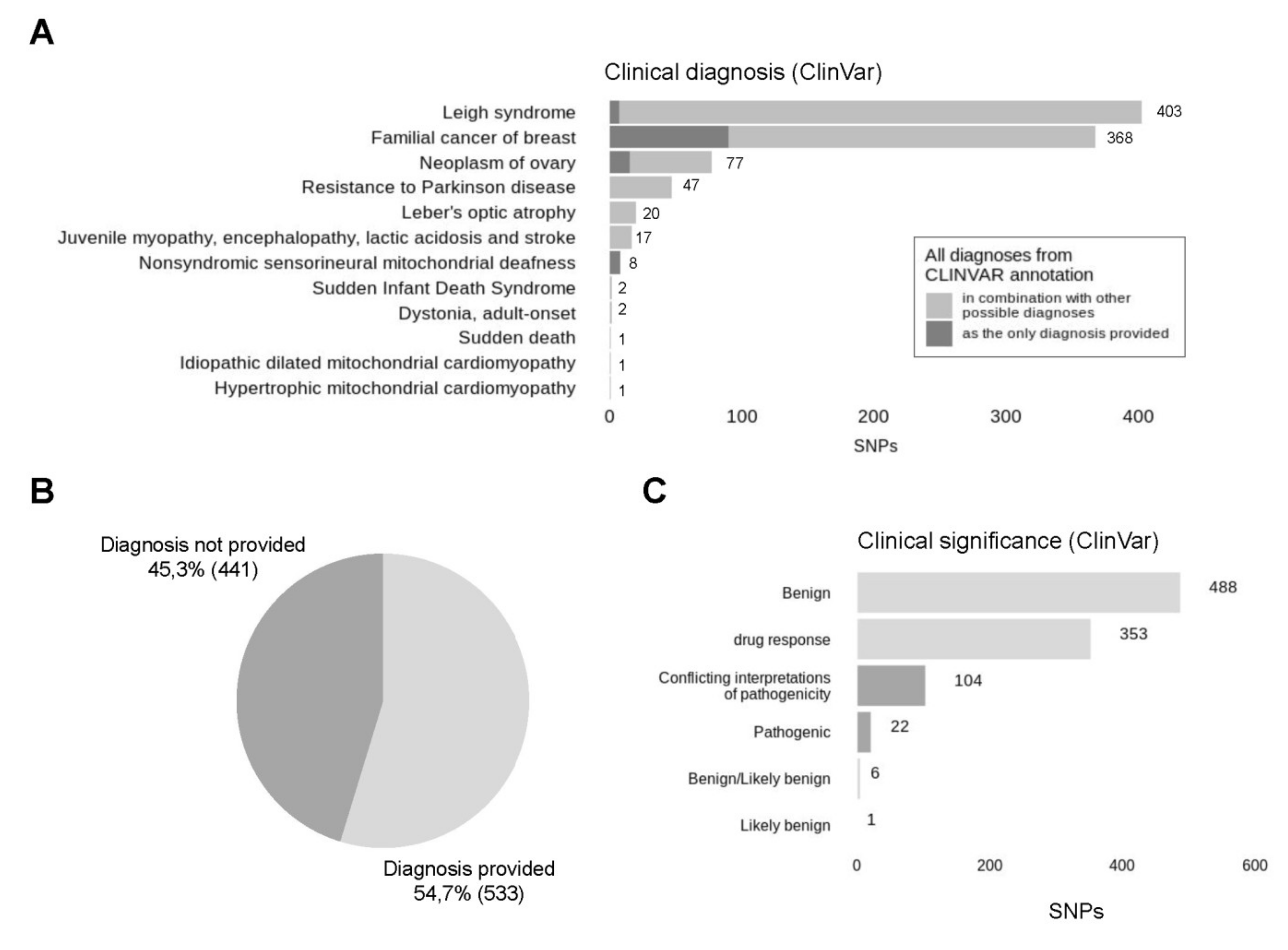
3.7. The Vast Majority of Called Variants Are Reported to Be Benign According to ClinVar Database
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lo, Y.M.D.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef]
- Suciu, I.D.; Toader, O.D.; Galeva, S.; Pop, L. Non-Invasive Prenatal Testing beyond Trisomies. J. Med. Life 2019, 12, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Carbone, I.F.; Conforti, A.; Picarelli, S.; Morano, D.; Alviggi, C.; Farina, A. Circulating Nucleic Acids in Maternal Plasma and Serum in Pregnancy Complications: Are They Really Useful in Clinical Practice? A Systematic Review. Mol. Diagn. Ther. 2020, 24, 409–431. [Google Scholar] [CrossRef] [PubMed]
- Amaral, L.M.; Sandrim, V.C.; Kutcher, M.E.; Spradley, F.T.; Cavalli, R.C.; Tanus-Santos, J.E.; Palei, A.C. Circulating Total Cell-Free DNA Levels Are Increased in Hypertensive Disorders of Pregnancy and Associated with Prohypertensive Factors and Adverse Clinical Outcomes. Int. J. Mol. Sci. 2021, 22, 564. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.M.; Quezada, M.S.; Bregant, B.; Ferraro, M.; Nicolaides, K.H. Implementation of maternal blood cell-free DNA testing in early screening for aneuploidies. Ultrasound Obstet. Gynecol. 2013, 42, 34–40. [Google Scholar] [CrossRef]
- Carbone, L.; Cariati, F.; Sarno, L.; Conforti, A.; Bagnulo, F.; Strina, I.; Pastore, L.; Maruotti, G.M.; Alviggi, C. Non-Invasive Prenatal Testing: Current Perspectives and Future Challenges. Genes 2020, 12, 15. [Google Scholar] [CrossRef]
- Bedei, I.; Wolter, A.; Weber, A.; Signore, F.; Axt-Fliedner, R. Chances and Challenges of New Genetic Screening Technologies (NIPT) in Prenatal Medicine from a Clinical Perspective: A Narrative Review. Genes 2021, 12, 501. [Google Scholar] [CrossRef]
- Newell, C.; Hume, S.; Greenway, S.C.; Podemski, L.; Shearer, J.; Khan, A. Plasma-derived cell-free mitochondrial DNA: A novel non-invasive methodology to identify mitochondrial DNA haplogroups in humans. Mol. Genet. Metab. 2018, 125, 332–337. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef]
- Hazkani-Covo, E.; Zeller, R.M.; Martin, W. Molecular Poltergeists: Mitochondrial DNA Copies (numts) in Sequenced Nuclear Genomes. PLoS Genet. 2010, 6, e1000834. [Google Scholar] [CrossRef]
- Goios, A.; Carvalho, A.; Amorim, A. Identifying NUMT contamination in mtDNA analyses. Forensic Sci. Int. Genet. Suppl. Ser. 2009, 2, 278–280. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The Release of Damage-Associated Molecular Patterns and Its Physiological Relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nat. Cell Biol. 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Bae, J.H.; Jo, S.I.; Kim, S.J.; Lee, J.M.; Jeong, J.H.; Kang, J.S.; Cho, N.-J.; Kim, S.S.; Lee, E.Y.; Moon, J.-S. Circulating Cell-Free mtDNA Contributes to AIM2 Inflammasome-Mediated Chronic Inflammation in Patients with Type 2 Diabetes. Cells 2019, 8, 328. [Google Scholar] [CrossRef]
- Agbor-Enoh, S.; Chan, J.L.; Singh, A.; Tunc, I.; Gorham, S.; Zhu, J.; Pirooznia, M.; Corcoran, P.C.; Thomas, M.L.; Lewis, B.G.; et al. Circulating cell-free DNA as a biomarker of tissue injury: Assessment in a cardiac xenotransplantation model. J. Hearth Lung Transplant. 2018, 37, 967–975. [Google Scholar] [CrossRef]
- Liu, J.; Cai, X.; Xie, L.; Tang, Y.; Cheng, J.; Wang, J.; Wang, L.; Gong, J. Circulating Cell Free Mitochondrial DNA is a Biomarker in the Development of Coronary Heart Disease in the Patients with Type 2 Diabetes. Clin. Lab. 2015, 59, 661–667. [Google Scholar] [CrossRef]
- Long, Y.; Zhang, Y.; Gong, Y.; Sun, R.; Su, L.; Lin, X.; Shen, A.; Zhou, J.; Caiji, Z.; Wang, X.; et al. Diagnosis of Sepsis with Cell-free DNA by Next-Generation Sequencing Technology in ICU Patients. Arch. Med. Res. 2016, 47, 365–371. [Google Scholar] [CrossRef]
- Podlesniy, P.; Figueiro-Silva, J.; Llado, A.; Antonell, A.; Sanchez-Valle, R.; Alcolea, D.; Lleo, A.; Molinuevo, J.L.; Serra, N.; Trullas, R. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann. Neurol. 2013, 74, 655–668. [Google Scholar] [CrossRef]
- Pyle, A.; Brennan, R.; Kurzawa-Akanbi, M.; Yarnall, A.J.; Thouin, A.; Mollenhauer, B.; Burn, D.J.; Chinnery, P.F.; Hudson, G. Reduced cerebrospinal fluid mitochondrial DNA is a biomarker for early-stage Parkinson’s disease. Ann. Neurol. 2015, 78, 1000–1004. [Google Scholar] [CrossRef]
- Cushen, S.C.; Sprouse, M.L.; Blessing, A.; Sun, J.; Jarvis, S.S.; Okada, Y.; Fu, Q.; Romero, S.A.; Phillips, N.R.; Goulopoulou, S. Cell-free mitochondrial DNA increases in maternal circulation during healthy pregnancy: A prospective, longitudinal study. Am. J. Physiol. Integr. Comp. Physiol. 2020, 318, R445–R452. [Google Scholar] [CrossRef]
- Van Boeckel, S.R.; Davidson, D.J.; Norman, J.E.; Stock, S.J. Cell-free fetal DNA and spontaneous preterm birth. Reproduction 2018, 155, R137–R145. [Google Scholar] [CrossRef]
- Busnelli, A.; Lattuada, D.; Ferrari, S.; Reschini, M.; Colciaghi, B.; Somigliana, E.; Fedele, L.; Ferrazzi, E. Mitochondrial DNA Copy Number in Peripheral Blood in the First Trimester of Pregnancy and Different Preeclampsia Clinical Phenotypes Development: A Pilot Study. Reprod. Sci. 2018, 26, 1054–1061. [Google Scholar] [CrossRef]
- Colleoni, F.; Lattuada, D.; Garretto, A.; Massari, M.; Mandò, C.; Somigliana, E.; Cetin, I. Maternal blood mitochondrial DNA content during normal and intrauterine growth restricted (IUGR) pregnancy. Am. J. Obstet. Gynecol. 2010, 203, 365.e1–365.e6. [Google Scholar] [CrossRef]
- Qiu, C.; Hevner, K.; Enquobahrie, D.A.; Williams, M.A. A case-control study of maternal blood mitochondrial DNA copy number and preeclampsia risk. Int. J. Mol. Epidemiol. Genet. 2012, 3, 237–244. [Google Scholar]
- Gilman-Sachs, A.; Dambaeva, S.; Garcia, M.D.S.; Hussein, Y.; Kwak-Kim, J.; Beaman, K. Inflammation induced preterm labor and birth. J. Reprod. Immunol. 2018, 129, 53–58. [Google Scholar] [CrossRef]
- Palacín, M.; Alvarez, V.; Martín, M.; Díaz, M.; Corao, A.I.; Alonso, B.; Díaz-Molina, B.; Lozano, I.; Avanzas, P.; Morís, C.; et al. Mitochondrial DNA and TFAM gene variation in early-onset myocardial infarction: Evidence for an association to haplogroup H. Mitochondrion 2011, 11, 176–181. [Google Scholar] [CrossRef]
- Smart, A.; Bolnick, D.A.; Tutton, R. Health and genetic ancestry testing: Time to bridge the gap. BMC Med. Genom. 2017, 10, 3. [Google Scholar] [CrossRef]
- Santoro, A.; Balbi, V.; Balducci, E.; Pirazzini, C.; Rosini, F.; Tavano, F.; Achilli, A.; Siviero, P.; Minicuci, N.; Bellavista, E.; et al. Evidence for Sub-Haplogroup H5 of Mitochondrial DNA as a Risk Factor for Late Onset Alzheimer’s Disease. PLoS ONE 2010, 5, e12037. [Google Scholar] [CrossRef]
- Ebner, S.; Lang, R.; Mueller, E.E.; Eder, W.; Oeller, M.; Moser, A.; Koller, J.; Paulweber, B.; Mayr, J.A.; Sperl, W.; et al. Mitochondrial Haplogroups, Control Region Polymorphisms and Malignant Melanoma: A Study in Middle European Caucasians. PLoS ONE 2011, 6, e27192. [Google Scholar] [CrossRef]
- Chinnery, P.F. Mitochondrial Disorders Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Thorburn, D.R.; Rahman, J.; Rahman, S. Mitochondrial DNA-Associated Leigh Syndrome and NARP. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J., 2nd; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Melton, T. Mitochondrial DNA Heteroplasmy. Forensic Sci. Rev. 2004, 16, 1–20. [Google Scholar]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Schneider, S.; Excoffier, L. Estimation of Past Demographic Parameters from the Distribution of Pairwise Differences When the Mutation Rates Vary Among Sites: Application to Human Mitochondrial DNA. Genetics 1999, 152, 1079–1089. [Google Scholar] [CrossRef]
- Nachman, M.W.; Crowell, S.L. Estimate of the Mutation Rate per Nucleotide in Humans. Genetics 2000, 156, 297–304. [Google Scholar] [CrossRef]
- Michikawa, Y.; Mazzucchelli, F.; Bresolin, N.; Scarlato, G.; Attardi, G. Aging-Dependent Large Accumulation of Point Mutations in the Human mtDNA Control Region for Replication. Science 1999, 286, 774–779. [Google Scholar] [CrossRef]
- Sharma, H.; Singh, A.; Sharma, C.; Jain, S.K.; Singh, N. Mutations in the mitochondrial DNA D-loop region are frequent in cervical cancer. Cancer Cell Int. 2005, 5, 34. [Google Scholar] [CrossRef][Green Version]
- Bahcall, O.G. mtDNA and genetic ancestry. Nat. Rev. Genet. 2015, 16, 128. [Google Scholar] [CrossRef]
- Hammer, M.F. A recent common ancestry for human Y chromosomes. Nat. Cell Biol. 1995, 378, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.L.; Goodloe, R.; Brown-Gentry, K.; Pendergrass, S.A.; Murdock, D.G.; Crawford, D.C. Characterization of mitochondrial haplogroups in a large population-based sample from the United States. Qual. Life Res. 2014, 133, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Pipek, O.A.; Medgyes-Horváth, A.; Dobos, L.; Stéger, J.; Szalai-Gindl, J.; Visontai, D.; Kaas, R.S.; Koopmans, M.; Hendriksen, R.S.; Aarestrup, F.M.; et al. Worldwide human mitochondrial haplogroup distribution from urban sewage. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Budis, J.; Gazdarica, J.; Radvanszky, J.; Harsanyova, M.; Gazdaricova, I.; Strieskova, L.; Frno, R.; Duris, F.; Minarik, G.; Sekelska, M.; et al. Non-invasive prenatal testing as a valuable source of population specific allelic frequencies. J. Biotechnol. 2019, 299, 72–78. [Google Scholar] [CrossRef]
- Pös, O.; Budis, J.; Kubiritova, Z.; Kucharik, M.; Duris, F.; Radvanszky, J.; Szemes, T. Identification of Structural Variation from NGS-Based Non-Invasive Prenatal Testing. Int. J. Mol. Sci. 2019, 20, 4403. [Google Scholar] [CrossRef]
- Keyser, C.; Hollard, C.; Gonzalez, A.; Fausser, J.-L.; Rivals, E.; Alexeev, A.N.; Riberon, A.; Crubézy, E.; Ludes, B. The ancient Yakuts: A population genetic enigma. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20130385. [Google Scholar] [CrossRef]
- Zlojutro, M.; Tarskaia, L.A.; Sorensen, M.; Snodgrass, J.J.; Leonard, W.R.; Crawford, M.H. Coalescent simulations of Yakut mtDNA variation suggest small founding population. Am. J. Phys. Anthropol. 2009, 139, 474–482. [Google Scholar] [CrossRef]
- Duda, P.; Zrzavý, J. Human population history revealed by a supertree approach. Sci. Rep. 2016, 6, 29890. [Google Scholar] [CrossRef]
- Wong, E.H.; Khrunin, A.; Nichols, L.; Pushkarev, D.; Khokhrin, D.; Verbenko, D.; Evgrafov, O.; Knowles, J.; Novembre, J.; Limborska, S.; et al. Reconstructing genetic history of Siberian and Northeastern European populations. Genome Res. 2016, 27, 1–14. [Google Scholar] [CrossRef]
- Kivisild, T. Maternal ancestry and population history from whole mitochondrial genomes. Investig. Genet. 2015, 6, 1–10. [Google Scholar] [CrossRef]
- McCormick, E.; Place, E.; Falk, M.J. Molecular Genetic Testing for Mitochondrial Disease: From One Generation to the Next. Neurotherapeutics 2012, 10, 251–261. [Google Scholar] [CrossRef]
- Wong, L.-J.C.; Scaglia, F.; Graham, B.H.; Craigen, W.J. Current molecular diagnostic algorithm for mitochondrial disorders. Mol. Genet. Metab. 2010, 100, 111–117. [Google Scholar] [CrossRef]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Lindenbaum, P. JVarkit: Java-Based Utilities for Bioinformatics; Institut du Thorax: Nantes, France, 2015. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Human hg19 UCSC Genome Browser. v399. Available online: http://genome.ucsc.edu/cgi-bin/hgTracks?hgsid=845883669_B1odJRHqogeppL15aTCtdmzGVmUl&position=&Submit=submit (accessed on 15 June 2020).
- Weissensteiner, H.; Pacher, D.; Kloss-Brandstätter, A.; Forer, L.; Specht, G.; Bandelt, H.-J.; Kronenberg, F.; Salas, A.; Schönherr, S. HaploGrep 2: Mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 2016, 44, W58–W63. [Google Scholar] [CrossRef]
- MITOMAP. Available online: https://www.mitomap.org/MITOMAP/TopVariants (accessed on 4 June 2020).
- Hidestrand, M.; Stokowski, R.; Song, K.; Oliphant, A.; Deavers, J.; Goetsch, M.; Simpson, P.; Kuhlman, R.; Ames, M.; Mitchell, M.; et al. Influence of Temperature during Transportation on Cell-Free DNA Analysis. Fetal Diagn. Ther. 2012, 31, 122–128. [Google Scholar] [CrossRef]
- Risberg, B.; Tsui, D.W.; Biggs, H.; de Almagro, A.R.-V.M.; Dawson, S.-J.; Hodgkin, C.; Jones, L.; Parkinson, C.; Piskorz, A.; Marass, F.; et al. Effects of Collection and Processing Procedures on Plasma Circulating Cell-Free DNA from Cancer Patients. J. Mol. Diagn. 2018, 20, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. DNA damage and apoptosis. Cell Death Differ. 2001, 8, 1047–1048. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Kowluru, R.A. Retinal Mitochondrial DNA Mismatch Repair in the Development of Diabetic Retinopathy, and Its Continued Progression After Termination of Hyperglycemia. Investig. Opthalmology Vis. Sci. 2014, 55, 6960–6967. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.; Moturi, S.; Angkachatchai, V.; Mueller, R.; DeSantis, G.; Boom, D.V.D.; Ehrich, M. Optimizing blood collection, transport and storage conditions for cell free DNA increases access to prenatal testing. Clin. Biochem. 2013, 46, 1099–1104. [Google Scholar] [CrossRef]
- Smith, M.; Lewis, K.M.; Holmes, A.; Visootsak, J. A Case of False Negative NIPT for Down Syndrome-Lessons Learned. Case Rep. Genet. 2014, 2014, 823504. [Google Scholar] [CrossRef]
- Yaron, Y. The implications of non-invasive prenatal testing failures: A review of an under-discussed phenomenon. Prenat. Diagn. 2016, 36, 391–396. [Google Scholar] [CrossRef]
- Hui, L.; Bianchi, D.W. Fetal fraction and noninvasive prenatal testing: What clinicians need to know. Prenat. Diagn. 2020, 40, 155–163. [Google Scholar] [CrossRef]
- Hartwig, T.S.; Ambye, L.; Sørensen, S.; Jørgensen, F.S. Discordant non-invasive prenatal testing (NIPT)—A systematic review. Prenat. Diagn. 2017, 37, 527–539. [Google Scholar] [CrossRef]
- Samura, O.; Okamoto, A. Causes of aberrant non-invasive prenatal testing for aneuploidy: A systematic review. Taiwan. J. Obstet. Gynecol. 2020, 59, 16–20. [Google Scholar] [CrossRef]
- Norwitz, E.R.; Levy, B. Noninvasive Prenatal Testing: The Future Is Now. Rev. Obstet. Gynecol. 2013, 6, 48–62. [Google Scholar]
- Ramos, A.; Santos, C.; Alvarez, L.; Nogués, R.; Aluja, M.P. Human mitochondrial DNA complete amplification and sequencing: A new validated primer set that prevents nuclear DNA sequences of mitochondrial origin co-amplification. Electrophoresis 2009, 30, 1587–1593. [Google Scholar] [CrossRef]
- Yao, Y.; Nishimura, M.; Murayama, K.; Kuranobu, N.; Tojo, S.; Beppu, M.; Ishige, T.; Itoga, S.; Tsuchida, S.; Mori, M.; et al. A simple method for sequencing the whole human mitochondrial genome directly from samples and its application to genetic testing. Sci. Rep. 2019, 9, 17411–17417. [Google Scholar] [CrossRef]
- Weerts, M.; Timmermans, E.; Van De Stolpe, A.; Vossen, R.; Anvar, S.; Foekens, J.; Sleijfer, S.; Martens, J. Tumor-Specific Mitochondrial DNA Variants Are Rarely Detected in Cell-Free DNA. Neoplasia 2018, 20, 687–696. [Google Scholar] [CrossRef]
- Benjamini, Y.; Speed, T.P. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012, 40, e72. [Google Scholar] [CrossRef]
- Lieber, T.; Jeedigunta, S.P.; Palozzi, J.M.; Lehmann, R.; Hurd, T.R. Mitochondrial fragmentation drives selective removal of deleterious mtDNA in the germline. Nat. Cell Biol. 2019, 570, 380–384. [Google Scholar] [CrossRef]
- Morozova, I.Y.; Naumova, O.Y.; Rychkov, S.Y.; Zhukova, O.V. Mitochondrial DNA Polymorphism in Russian Population form Five Oblasts of the European Part of Russia. Russ. J. Genet. 2005, 41, 1040–1045. [Google Scholar] [CrossRef]
- Malyarchuk, B.A.; Derenko, M.V.; Grzybowski, T.; Lunkina, A.; Czarny, J.; Rychkov, S.; Morozova, I.; Denisova, G.; Miścicka-Sliwka, D. Differentiation of Mitochondrial DNA and Y Chromosomes in Russian Populations. Hum. Biol. 2004, 76, 877–900. [Google Scholar] [CrossRef]
- Puzyrev, V.P.; Stepanov, V.A.; Golubenko, M.V.; Puzyrev, K.V.; Maksimova, N.R.; Kharkov, V.N.; Spiridonova, M.G.; Nogovitsyna, A.N. MtDNA and Y-chromosome lineages in the Yakut population. Russ. J. Genet. 2003, 39, 975–981. [Google Scholar] [CrossRef]
- Kehdy, F.S.G.; Gouveia, M.H.; Machado, M.; Magalhães, W.C.S.; Horimoto, A.R.; Horta, B.L.; Moreira, R.G.; Leal, T.P.; Scliar, M.O.; Soares-Souza, G.B.; et al. Origin and dynamics of admixture in Brazilians and its effect on the pattern of deleterious mutations. Proc. Natl. Acad. Sci. USA 2015, 112, 8696–8701. [Google Scholar] [CrossRef] [PubMed]
- Shoubridge, E.A. Mitochondrial DNA segregation in the developing embryo. Hum. Reprod. 2000, 15, 229–234. [Google Scholar] [CrossRef]
- Zhang, P.; Samuels, D.C.; Wang, J.; Zhao, S.; Shyr, Y.; Guo, Y. Mitochondria single nucleotide variation across six blood cell types. Mitochondrion 2016, 28. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Number of Variants | Indels | SNPs | |
|---|---|---|---|
| Total | 32,962 | 21,416 | 11,546 |
| Depth filtering | 22,017 | 17,035 | 4982 |
| Homopolymer filtering (≥4) | 14,681 | 10,479 | 4202 |
| Homopolymer filtering (≥3) | 8969 | 6450 | 2519 |
| mtDNA Variant | Patients |
|---|---|
| m.15301G>A | 43 (6.7%) |
| m.489T>C | 36 (5.6%) |
| m.10400C>T | 36 (5.6%) |
| m.14783T>C | 34 (5.3%) |
| m.15452C>A | 29 (4.5%) |
| mtDNA Variant | Patients |
|---|---|
| m.9906delG | 81 (12.6%) |
| m.10151delA | 44 (6.8%) |
| m.9916delC | 38 (5.6%) |
| m.2193delT | 28 (4.3%) |
| m.9808insT | 27 (4.2%) |
| mtDNA Variant | rs ID | ClinVar ID | Clinical Significance | Diagnosis (ClinVar) | Number of Patients |
|---|---|---|---|---|---|
| m.15301G>A | 193302991 | 140591 | Conflicting interpretations of pathogenicity | Familial cancer of breast | 43 (6.7%) |
| m.14783T>C | 193302982 | 140588 | Conflicting interpretations of pathogenicity | Familial cancer of breast | 34 (5.3%) |
| m.15452C>A | 193302994 | 143925 | Benign | Neoplasm of ovary/ Leigh syndrome | 29 (4.5%) |
| m.3010G>A | 3928306 | 441149 | Drug response | Not provided | 28 (4.3%) |
| m.13708G>A | 28359178 | 9696 | Benign | Leber’s optic atrophy/Leigh syndrome | 10 (1.6%) |
| DNA Seq Variant | Central (%) | Northwestern (%) | Volga-Ural (%) | Yakutia (%) | Others (%) | Number of Patients |
|---|---|---|---|---|---|---|
| m.15301G>A | 1.81 | 8.00 | 0.0 | 79.17 | 0.00 | 54 (8.4%) |
| m.10400C>T | 0.00 | 2.67 | 2.5 | 75.00 | 0.00 | 41 (6.4%) |
| m.12704TC>T | 1.36 | 3.33 | 0.0 | 47.92 | 0.00 | 31 (4.8%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morshneva, A.; Kozyulina, P.; Vashukova, E.; Tarasenko, O.; Dvoynova, N.; Chentsova, A.; Talantova, O.; Koroteev, A.; Ivanov, D.; Serebryakova, E.; et al. Pilot Screening of Cell-Free mtDNA in NIPT: Quality Control, Variant Calling, and Haplogroup Determination. Genes 2021, 12, 743. https://doi.org/10.3390/genes12050743
Morshneva A, Kozyulina P, Vashukova E, Tarasenko O, Dvoynova N, Chentsova A, Talantova O, Koroteev A, Ivanov D, Serebryakova E, et al. Pilot Screening of Cell-Free mtDNA in NIPT: Quality Control, Variant Calling, and Haplogroup Determination. Genes. 2021; 12(5):743. https://doi.org/10.3390/genes12050743
Chicago/Turabian StyleMorshneva, Alisa, Polina Kozyulina, Elena Vashukova, Olga Tarasenko, Natalia Dvoynova, Anastasia Chentsova, Olga Talantova, Alexander Koroteev, Dmitrii Ivanov, Elena Serebryakova, and et al. 2021. "Pilot Screening of Cell-Free mtDNA in NIPT: Quality Control, Variant Calling, and Haplogroup Determination" Genes 12, no. 5: 743. https://doi.org/10.3390/genes12050743
APA StyleMorshneva, A., Kozyulina, P., Vashukova, E., Tarasenko, O., Dvoynova, N., Chentsova, A., Talantova, O., Koroteev, A., Ivanov, D., Serebryakova, E., Ivashchenko, T., Sukhomyasova, A., Maksimova, N., Bespalova, O., Kogan, I., Baranov, V., & Glotov, A. (2021). Pilot Screening of Cell-Free mtDNA in NIPT: Quality Control, Variant Calling, and Haplogroup Determination. Genes, 12(5), 743. https://doi.org/10.3390/genes12050743