
New Insights into the Therapeutic Applications of CRISPR/Cas9 Genome Editing in Breast Cancer


Abstract
1. Introduction
2. CRISPR/Cas9 System
3. Potential CRISPR/Cas9 Targets
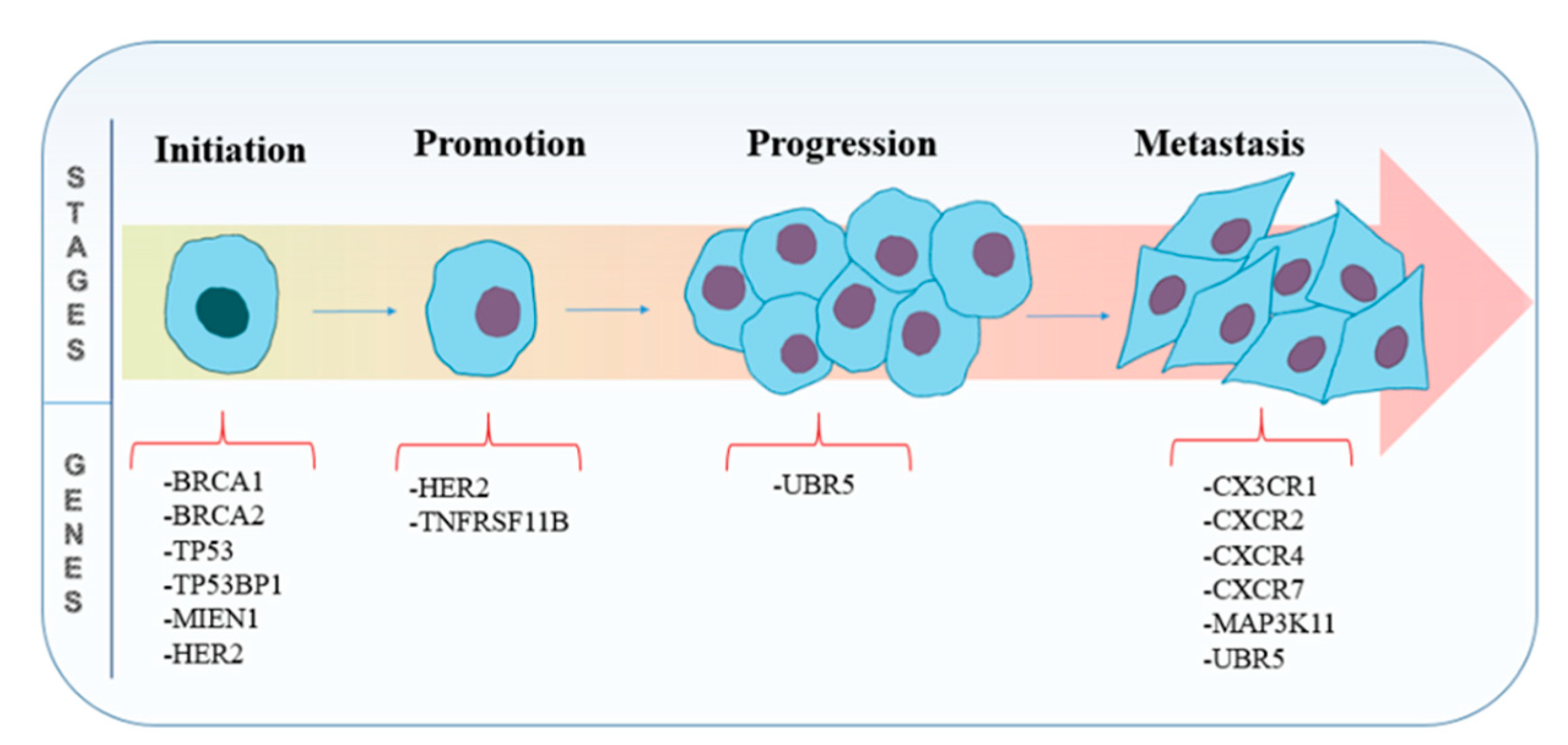
3.1. Genes Involved in Tumorigenesis
3.1.1. BRCA1
3.1.2. BRCA2
3.1.3. HER2
3.1.4. TP53
3.1.5. TP53PB1
3.1.6. MKI67
3.2. Genes Involved in Metastasis
3.2.1. MIEN1
3.2.2. CX3CR1
3.2.3. CXCR2
3.2.4. CXCR4 and CXCR7
3.2.5. MAP3K11
3.2.6. TNFRSF11B
3.2.7. UBR5
4. Delivery Methods for CRISPR/Cas9
4.1. Viral Delivery
4.1.1. Adenoviruses (AdVs)
4.1.2. Lentiviruses (LVs)
4.1.3. Adeno-Associated Viruses (AAVs)
4.2. Non-Viral Delivery
4.2.1. Electroporation
4.2.2. Microinjection
4.2.3. Calcium Phosphate Delivery
4.2.4. Lipid-Mediated Delivery
4.3. Targeted Delivery
5. Clinical Trials
6. Implications for the Future
6.1. Precision Genome Editing
6.2. Mapping Genetic Interactions
6.3. Synthetic Lethality Screens
7. Limitations
7.1. Off-Target Effects
7.2. Protospacer Adjacent Motif Requirement
7.3. Delivery
7.4. Ethical Consideration
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; de la Garza Salazar, J.; Pritchard, K.; Amadori, D.; Haidinger, R.; Hudis, C.A.; Khaled, H.; Liu, M.-C.; Martin, M.; Namer, M.; et al. The Global Breast Cancer Burden: Variations in Epidemiology and Survival. Clin. Breast Cancer 2005, 6, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR—Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef] [PubMed]
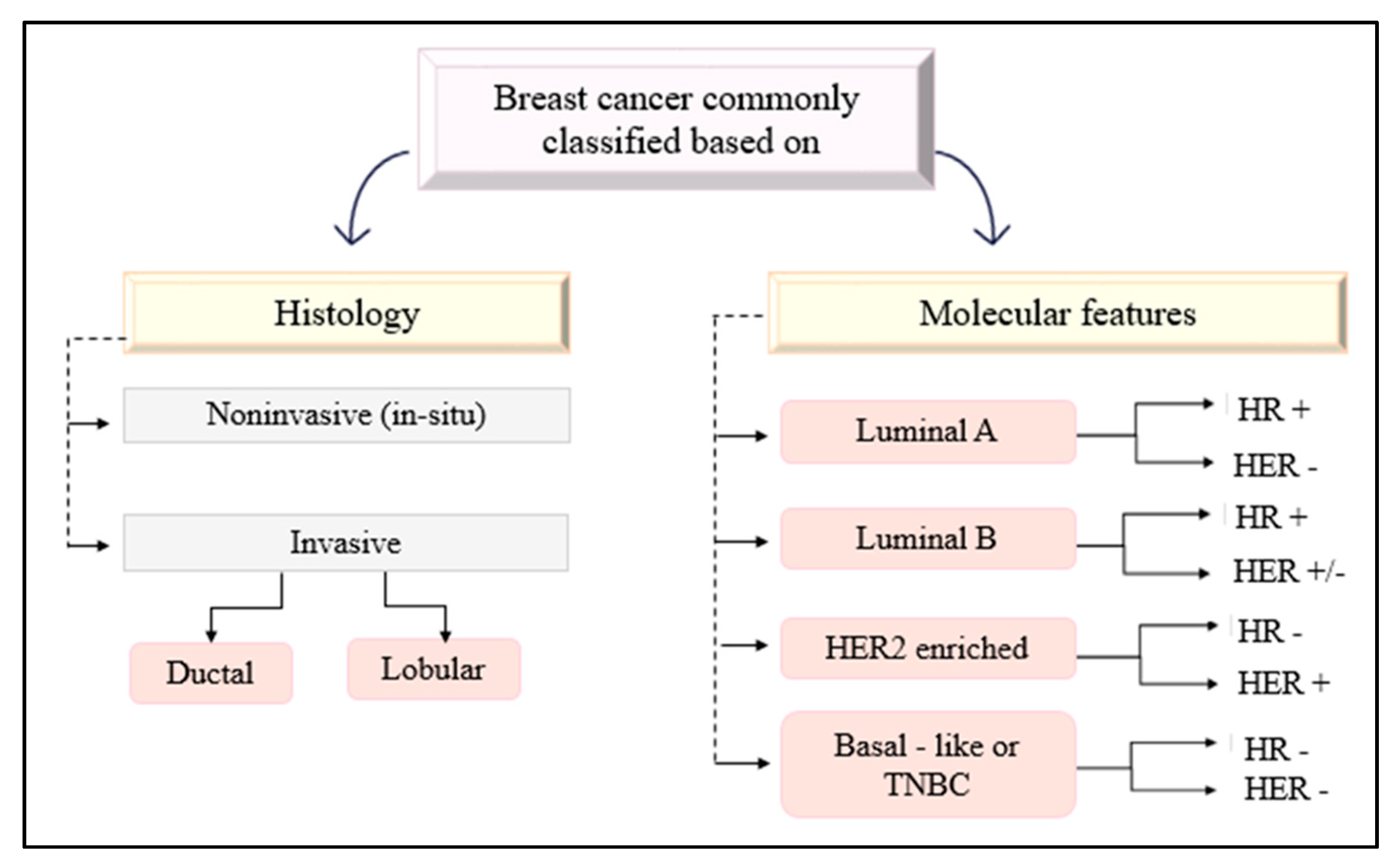
- Makki, J. Diversity of Breast Carcinoma: Histological Subtypes and Clinical Relevance. Clin. Med. Insights Pathol. 2015, 8, CPath.S31563. [Google Scholar] [CrossRef]
- Akram, M.; Iqbal, M.; Daniyal, M.; Khan, A.U. Awareness and current knowledge of breast cancer. Biol. Res. 2017, 50, 33. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, L.; Huang, X. Genome modification by CRISPR/Cas9. FEBS J. 2014, 281, 5186–5193. [Google Scholar] [CrossRef]
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The Biology of CRISPR-Cas: Backward and Forward. Cell 2018, 172, 1239–1259. [Google Scholar] [CrossRef]
- Mojica, F.J.M.; Diez-Villasenor, C.; Garcia-Martinez, J.; Soria, E. Intervening Sequences of Regularly Spaced Prokaryotic Repeats Derive from Foreign Genetic Elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Arnedos, M.; Vicier, C.; Loi, S.; Lefebvre, C.; Michiels, S.; Bonnefoi, H.; Andre, F. Precision medicine for metastatic breast cancer—limitations and solutions. Nat. Rev. Clin. Oncol. 2015, 12, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.H. Genome-Editing Technologies: Concept, Pros, and Cons of Various Genome-Editing Techniques and Bioethical Concerns for Clinical Application. Mol. Ther. Nucleic Acids 2019, 16, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Salsman, J.; Dellaire, G. Precision genome editing in the CRISPR era. Biochem. Cell Biol. 2017, 95, 187–201. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Adikaram, P.; Pandey, M.; Genis, A.; Simonds, W.F. Optimization of genome editing through CRISPR-Cas9 engineering. Bioengineered 2016, 7, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Riehle, M.M.; Bennett, A.F.; Long, A.D. Genetic architecture of thermal adaptation in Escherichia coli. Proc. Natl. Acad. Sci. USA 2001, 98, 525–530. [Google Scholar] [CrossRef]
- DeBoy, R.T.; Mongodin, E.F.; Emerson, J.B.; Nelson, K.E. Chromosome Evolution in the Thermotogales: Large-Scale Inversions and Strain Diversification of CRISPR Sequences. J. Bacteriol. 2006, 188, 2364–2374. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; van Embden, J.D.A.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef]
- Mojica, F.J.M.; Diez-Villasenor, C.; Soria, E.; Juez, G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol. 2000, 36, 244–246. [Google Scholar] [CrossRef]
- Pourcel, C.; Salvignol, G.; Vergnaud, G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 2005, 151, 653–663. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR–Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR—Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Barrangou, R. CRISPR-Cas systems and RNA-guided interference: CRISPR-Cas systems and RNA-guided interference. WIREs RNA 2013, 4, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Bhaya, D.; Davison, M.; Barrangou, R. CRISPR-Cas Systems in Bacteria and Archaea: Versatile Small RNAs for Adaptive Defense and Regulation. Annu. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Yosef, I.; Goren, M.G.; Qimron, U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012, 40, 5569–5576. [Google Scholar] [CrossRef]
- Nuñez, J.K.; Kranzusch, P.J.; Noeske, J.; Wright, A.V.; Davies, C.W.; Doudna, J.A. Cas1–Cas2 complex formation mediates spacer acquisition during CRISPR–Cas adaptive immunity. Nat. Struct. Mol. Biol. 2014, 21, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Heler, R.; Samai, P.; Modell, J.W.; Weiner, C.; Goldberg, G.W.; Bikard, D.; Marraffini, L.A. Cas9 specifies functional viral targets during CRISPR–Cas adaptation. Nature 2015, 519, 199–202. [Google Scholar] [CrossRef]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef]
- Thurtle-Schmidt, D.M.; Lo, T.-W. Molecular biology at the cutting edge: A review on CRISPR/CAS9 gene editing for undergraduates: A Review on CRISPR/CAS9 Gene Editing for Undergraduates. Biochem. Mol. Biol. Educ. 2018, 46, 195–205. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 Endonucleases Reveal RNA-Mediated Conformational Activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rodriguez, D.; Ramirez-Solis, R.; Garza-Elizondo, M.; Garza-Rodriguez, M.; Barrera-Saldana, H. Genome editing: A perspective on the application of CRISPR/Cas9 to study human diseases (Review). Int. J. Mol. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Biagioni, A.; Chillà, A.; Andreucci, E.; Laurenzana, A.; Margheri, F.; Peppicelli, S.; Del Rosso, M.; Fibbi, G. Type II CRISPR/Cas9 approach in the oncological therapy. J. Exp. Clin. Cancer Res. 2017, 36, 80. [Google Scholar] [CrossRef]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef]
- Wyman, C.; Kanaar, R. DNA Double-Strand Break Repair: All’s Well that Ends Well. Annu. Rev. Genet. 2006, 40, 363–383. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef]
- Martinez-Lage, M.; Puig-Serra, P.; Menendez, P.; Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges. Biomedicines 2018, 6, 105. [Google Scholar] [CrossRef]
- Petrucelli, N.; Daly, M.B.; Feldman, G.L. Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genet. Med. 2010, 12, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.Y.; Sy, S.M.H.; Chen, J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 2010, 11, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Rajagopal, P.; Hoyt, D.W.; King, M.-C.; Klevit, R.E. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 2001, 8, 833–837. [Google Scholar] [CrossRef]
- Brzovic, P.S.; Keeffe, J.R.; Nishikawa, H.; Miyamoto, K.; Fox, D.; Fukuda, M.; Ohta, T.; Klevit, R. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc. Natl. Acad. Sci. USA 2003, 100, 5646–5651. [Google Scholar] [CrossRef] [PubMed]
- Yamane, K.; Katayama, E.; Tsuruo, T. The BRCT Regions of Tumor Suppressor BRCA1 and of XRCC1 Show DNA End Binding Activity with a Multimerizing Feature. Biochem. Biophys. Res. Commun. 2000, 279, 678–684. [Google Scholar] [CrossRef]
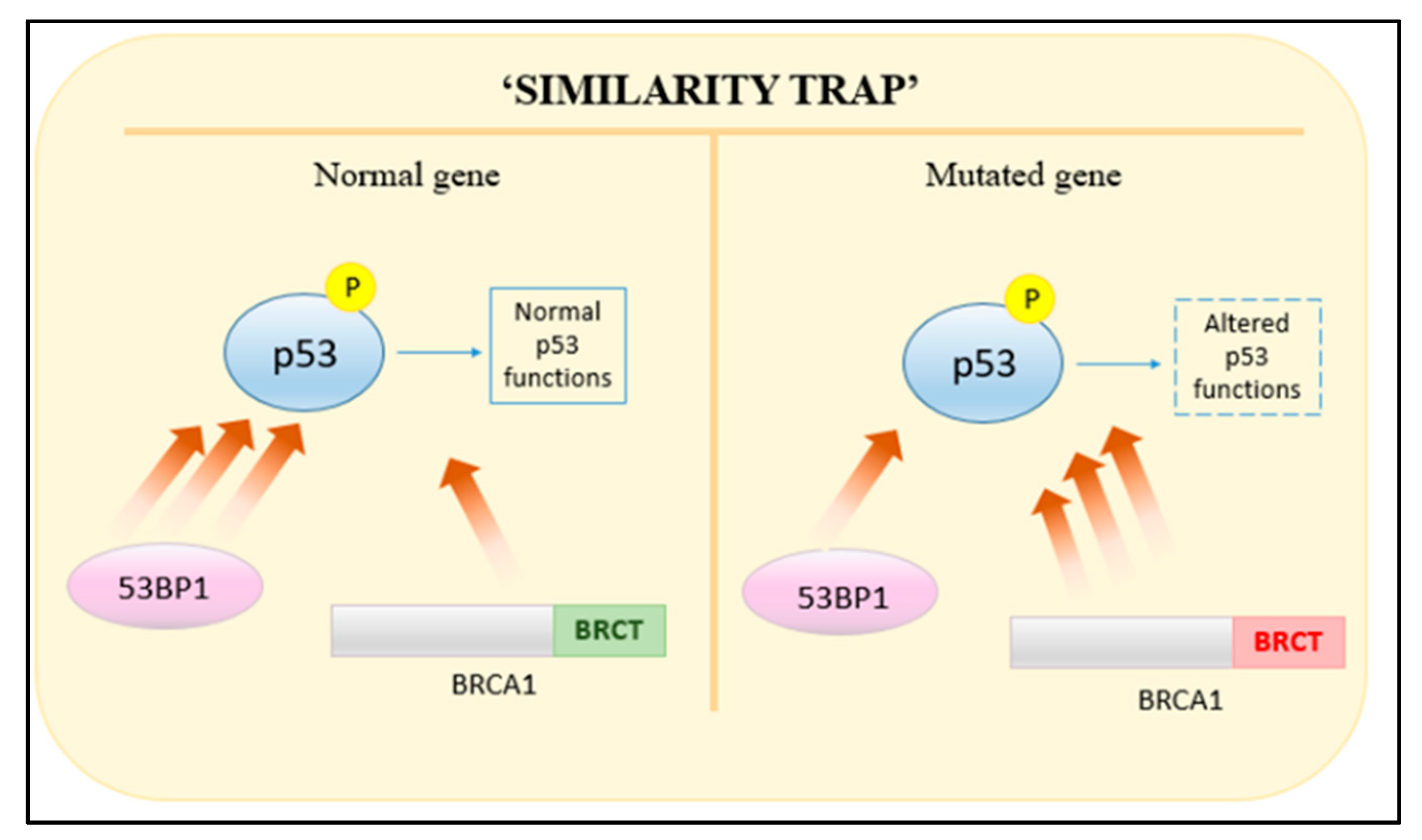
- Liu, J.; Pan, Y.; Ma, B.; Nussinov, R. “Similarity Trap” in Protein-Protein Interactions Could Be Carcinogenic: Simulations of p53 Core Domain Complexed with 53BP1 and BRCA1 BRCT Domains. Structure 2006, 14, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Kweon, J.; Jang, A.-H.; Shin, H.R.; See, J.-E.; Lee, W.; Lee, J.W.; Chang, S.; Kim, K.; Kim, Y. A CRISPR-based base-editing screen for the functional assessment of BRCA1 variants. Oncogene 2020, 39, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Corso, G.; Feroce, I.; Intra, M.; Toesca, A.; Magnoni, F.; Sargenti, M.; Naninato, P.; Caldarella, P.; Pagani, G.; Vento, A.; et al. BRCA1/2 germline missense mutations: A systematic review. Eur. J. Cancer Prev. 2018, 27, 279–286. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef]
- Suwaki, N.; Klare, K.; Tarsounas, M. RAD51 paralogs: Roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin. Cell Dev. Biol. 2011, 22, 898–905. [Google Scholar] [CrossRef]
- Wong, M.W.; Nordfors, C.; Mossman, D.; Pecenpetelovska, G.; Avery-Kiejda, K.A.; Talseth-Palmer, B.; Bowden, N.A.; Scott, R.J. BRIP1, PALB2, and RAD51C mutation analysis reveals their relative importance as genetic susceptibility factors for breast cancer. Breast Cancer Res. Treat. 2011, 127, 853–859. [Google Scholar] [CrossRef]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Jasin, M. BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nat. Commun. 2017, 8, 525. [Google Scholar] [CrossRef]
- Slamon, D.; Clark, G.; Wong, S.; Levin, W.; Ullrich, A.; McGuire, W. Human Breast Cancer: Correlation of Relapse and Survival with Amplification of the HER-2/neu Oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, W. CRISPR-mediated targeting of HER2 inhibits cell proliferation through a dominant negative mutation. Cancer Lett. 2017, 385, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, C.; Schiff, R. HER2: Biology, detection, and clinical implications. Arch. Pathol. Lab. Med. 2011, 135, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y. Biology of HER2 and Its Importance in Breast Cancer. Oncology 2001, 61, 1–13. [Google Scholar] [CrossRef]
- Moasser, M.M. The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef]
- Home—My Cancer Genome. Available online: https://www.mycancergenome.org/ (accessed on 27 November 2020).
- Welcome—IARC TP53 Database. Available online: https://p53.iarc.fr/ (accessed on 27 November 2020).
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef]
- Levine, A.J.; Berger, S.L. The interplay between epigenetic changes and the p53 protein in stem cells. Genes Dev. 2017, 31, 1195–1201. [Google Scholar] [CrossRef]
- Metastasis Has Multiple Origins and Occurs Early in Tumorigenesis. Cancer Discov. 2020, 10, 903. [CrossRef]
- Belotserkovskaya, R.; Raga Gil, E.; Lawrence, N.; Butler, R.; Clifford, G.; Wilson, M.D.; Jackson, S.P. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nat. Commun. 2020, 11, 819. [Google Scholar] [CrossRef]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 Cellular and Clinical Functions by a Nuclear Partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Ripperger, T.; Gadzicki, D.; Meindl, A.; Schlegelberger, B. Breast cancer susceptibility: Current knowledge and implications for genetic counselling. Eur. J. Hum. Genet. 2009, 17, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Wiltshire, T.; Ducy, M.; Foo, T.K.; Hu, C.; Lee, K.Y.; Belur Nagaraj, A.; Rodrigue, A.; Gomes, T.T.; Simard, J.; Monteiro, A.N.A.; et al. Functional characterization of 84 PALB2 variants of uncertain significance. Genet. Med. 2020, 22, 622–632. [Google Scholar] [CrossRef]
- Cidado, J.; Wong, H.Y.; Rosen, D.M.; Cimino-Mathews, A.; Garay, J.P.; Fessler, A.G.; Rasheed, Z.A.; Hicks, J.; Cochran, R.L.; Croessmann, S.; et al. Ki-67 is required for maintenance of cancer stem cells but not cell proliferation. Oncotarget 2016, 7, 6281–6293. [Google Scholar] [CrossRef] [PubMed]
- Castéra, L.; Harter, V.; Muller, E.; Krieger, S.; Goardon, N.; Ricou, A.; Rousselin, A.; Paimparay, G.; Legros, A.; Bruet, O.; et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet. Med. 2018, 20, 1677–1686. [Google Scholar] [CrossRef]
- Chapman, J.R.; Sossick, A.J.; Boulton, S.J.; Jackson, S.P. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012, 125, 3529–3534. [Google Scholar] [CrossRef]
- Endl, E.; Gerdes, J. The Ki-67 Protein: Fascinating Forms and an Unknown Function. Exp. Cell Res. 2000, 257, 231–237. [Google Scholar] [CrossRef]
- Cuzick, J.; Dowsett, M.; Pineda, S.; Wale, C.; Salter, J.; Quinn, E.; Zabaglo, L.; Mallon, E.; Green, A.R.; Ellis, I.O.; et al. Prognostic Value of a Combined Estrogen Receptor, Progesterone Receptor, Ki-67, and Human Epidermal Growth Factor Receptor 2 Immunohistochemical Score and Comparison With the Genomic Health Recurrence Score in Early Breast Cancer. JCO 2011, 29, 4273–4278. [Google Scholar] [CrossRef]
- Cuylen, S.; Blaukopf, C.; Politi, A.Z.; Müller-Reichert, T.; Neumann, B.; Poser, I.; Ellenberg, J.; Hyman, A.A.; Gerlich, D.W. Ki-67 acts as a biological surfactant to disperse mitotic chromosomes. Nature 2016, 535, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Sobecki, M.; Mrouj, K.; Camasses, A.; Parisis, N.; Nicolas, E.; Llères, D.; Gerbe, F.; Prieto, S.; Krasinska, L.; David, A.; et al. The cell proliferation antigen Ki-67 organises heterochromatin. eLife 2016, 5, e13722. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Natsume, T.; Kanemaki, M.T.; Imamoto, N. Perichromosomal protein Ki67 supports mitotic chromosome architecture. Genes Cells 2016, 21, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-Cycle Regulation Accounts for Variability in Ki-67 Expression Levels. Cancer Res. 2017, 77, 2722–2734. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.; Min, M.; Yang, C.; Tian, C.; Gookin, S.; Carter, D.; Spencer, S.L. Ki67 is a Graded Rather than a Binary Marker of Proliferation versus Quiescence. Cell Rep. 2018, 24, 1105–1112.e5. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.L.; Kowalski, H.M.; Blazar, B.A.; Fligiel, Z.; Vogel, R.; Heppner, G.H. Heterogeneity of tumor cells from a single mouse mammary tumor. Cancer Res. 1978, 38, 3174–3181. [Google Scholar] [PubMed]
- Mrouj, K.; Singh, P.; Sobecki, M.; Dubra, G.; Al Ghoul, E.; Aznar, A.; Prieto, S.; Pirot, N.; Bernex, F.; Bordignon, B.; et al. Ki-67 promotes sequential stages of tumourigenesis by enabling cellular plasticity. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Redig, A.J.; McAllister, S.S. Breast cancer as a systemic disease: A view of metastasis. J. Intern. Med. 2013, 274, 113–126. [Google Scholar] [CrossRef]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Ocaña, O.H.; Córcoles, R.; Fabra, Á.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic Colonization Requires the Repression of the Epithelial-Mesenchymal Transition Inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A. Molecular envoys pave the way for pancreatic cancer to invade the liver. Nature 2019, 567, 181–182. [Google Scholar] [CrossRef]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Nieto, M.A. The Ins and Outs of the Epithelial to Mesenchymal Transition in Health and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, P.P.; Gupta, S.; Singh, A.K.; Kumar, S. Emerging Role of Migration and Invasion Enhancer 1 (MIEN1) in Cancer Progression and Metastasis. Front. Oncol. 2019, 9, 868. [Google Scholar] [CrossRef] [PubMed]
- Kpetemey, M.; Dasgupta, S.; Rajendiran, S.; Das, S.; Gibbs, L.D.; Shetty, P.; Gryczynski, Z.; Vishwanatha, J.K. MIEN1, a novel interactor of Annexin A2, promotes tumor cell migration by enhancing AnxA2 cell surface expression. Mol. Cancer 2015, 14, 156. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Wasson, L.M.; Rauniyar, N.; Prokai, L.; Borejdo, J.; Vishwanatha, J.K. Novel gene C17orf37 in 17q12 amplicon promotes migration and invasion of prostate cancer cells. Oncogene 2009, 28, 2860–2872. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Cushman, I.; Kpetemey, M.; Casey, P.J.; Vishwanatha, J.K. Prenylated C17orf37 Induces Filopodia Formation to Promote Cell Migration and Metastasis. J. Biol. Chem. 2011, 286, 25935–25946. [Google Scholar] [CrossRef]
- Van Treuren, T.; Vishwanatha, J.K. CRISPR deletion of MIEN1 in breast cancer cells. PLoS ONE 2018, 13, e0204976. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Zhang, Y.; Jernigan, D.L.; Feng, X.; Yan, J.; Garcia, F.U.; Meucci, O.; Salvino, J.M.; Fatatis, A. Novel Small-Molecule CX3CR1 Antagonist Impairs Metastatic Seeding and Colonization of Breast Cancer Cells. Mol. Cancer Res. 2016, 14, 518–527. [Google Scholar] [CrossRef]
- Bazan, J.F.; Bacon, K.B.; Hardiman, G.; Wang, W.; Soo, K.; Rossi, D.; Greaves, D.R.; Zlotnik, A.; Schall, T.J. A new class of membrane-bound chemokine with a CX3C motif. Nature 1997, 385, 640–644. [Google Scholar] [CrossRef]
- Onitilo, A.A.; Engel, J.M.; Greenlee, R.T.; Mukesh, B.N. Breast Cancer Subtypes Based on ER/PR and Her2 Expression: Comparison of Clinicopathologic Features and Survival. Clin. Med. Res. 2009, 7, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Gruber, I.; Fehm, T.; Taran, F.A.; Wallwiener, M.; Hahn, M.; Wallwiener, D.; Krawzyck, N.; Hoffmann, J.; Hartkopf, A.D. Disseminated tumor cells as a monitoring tool for adjuvant therapy in patients with primary breast cancer. Breast Cancer Res. Treat. 2014, 144, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.N.; Chen, S.-C.; Sullivan, L.M.; Manfra, D.J.; Wiekowski, M.T.; Prosser, D.M.; Vassileva, G.; Lira, S.A. Generation and Analysis of Mice Lacking the Chemokine Fractalkine. Mol. Cell. Biol. 2001, 21, 3159–3165. [Google Scholar] [CrossRef]
- Jamieson-Gladney, W.L.; Zhang, Y.; Fong, A.M.; Meucci, O.; Fatatis, A. The chemokine receptor CX3CR1 is directly involved in the arrest of breast cancer cells to the skeleton. Breast Cancer Res. 2011, 13, R91. [Google Scholar] [CrossRef] [PubMed]
- Xie, K. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev. 2001, 12, 375–391. [Google Scholar] [CrossRef]
- Long, X.; Ye, Y.; Zhang, L.; Liu, P.; Yu, W.; Wei, F.; Ren, X.; Yu, J. IL-8, a novel messenger to cross-link inflammation and tumor EMT via autocrine and paracrine pathways (Review). Int. J. Oncol. 2016, 48, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Stillie, R.; Farooq, S.M.; Gordon, J.R.; Stadnyk, A.W. The functional significance behind expressing two IL-8 receptor types on PMN. J. Leukoc. Biol. 2009, 86, 529–543. [Google Scholar] [CrossRef]
- Wuyts, A.; Van Osselaer, N.; Haelens, A.; Samson, I.; Herdewijn, P.; Ben-Baruch, A.; Oppenheim, J.J.; Proost, P.; Van Damme, J. Characterization of Synthetic Human Granulocyte Chemotactic Protein 2: Usage of Chemokine Receptors CXCR1 and CXCR2 and in Vivo Inflammatory Properties. Biochemistry 1997, 36, 2716–2723. [Google Scholar] [CrossRef]
- Saintigny, P.; Massarelli, E.; Lin, S.; Ahn, Y.-H.; Chen, Y.; Goswami, S.; Erez, B.; O’Reilly, M.S.; Liu, D.; Lee, J.J.; et al. CXCR2 Expression in Tumor Cells Is a Poor Prognostic Factor and Promotes Invasion and Metastasis in Lung Adenocarcinoma. Cancer Res. 2013, 73, 571–582. [Google Scholar] [CrossRef]
- Jin, K.; Pandey, N.B.; Popel, A.S. Crosstalk between stromal components and tumor cells of TNBC via secreted factors enhances tumor growth and metastasis. Oncotarget 2017, 8, 60210–60222. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zeng, C.; Li, P.; Qian, L.; Ding, B.; Huang, L.; Li, G.; Jiang, H.; Gong, N.; Wu, W. Impact of CXCR4 and CXCR7 knockout by CRISPR/Cas9 on the function of triple-negative breast cancer cells. OncoTargets Ther. 2019, 12, 3849–3858. [Google Scholar] [CrossRef] [PubMed]
- Ganju, R.K.; Brubaker, S.A.; Meyer, J.; Dutt, P.; Yang, Y.; Qin, S.; Newman, W.; Groopman, J.E. The α-Chemokine, Stromal Cell-derived Factor-1α, Binds to the Transmembrane G-protein-coupled CXCR-4 Receptor and Activates Multiple Signal Transduction Pathways. J. Biol. Chem. 1998, 273, 23169–23175. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Luker, K.E.; Summers, B.C.; Berahovich, R.; Bhojani, M.S.; Rehemtulla, A.; Kleer, C.G.; Essner, J.J.; Nasevicius, A.; Luker, G.D.; et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc. Natl. Acad. Sci. USA 2007, 104, 15735–15740. [Google Scholar] [CrossRef]
- Wang, J.; Shiozawa, Y.; Wang, J.; Wang, Y.; Jung, Y.; Pienta, K.J.; Mehra, R.; Loberg, R.; Taichman, R.S. The Role of CXCR7/RDC1 as a Chemokine Receptor for CXCL12/SDF-1 in Prostate Cancer. J. Biol. Chem. 2008, 283, 4283–4294. [Google Scholar] [CrossRef]
- Zheng, K.; Li, H.-Y.; Su, X.-L.; Wang, X.-Y.; Tian, T.; Li, F.; Ren, G.-S. Chemokine receptor CXCR7 regulates the invasion, angiogenesis and tumor growth of human hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2010, 29, 31. [Google Scholar] [CrossRef]
- Burns, J.M.; Summers, B.C.; Wang, Y.; Melikian, A.; Berahovich, R.; Miao, Z.; Penfold, M.E.T.; Sunshine, M.J.; Littman, D.R.; Kuo, C.J.; et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 2006, 203, 2201–2213. [Google Scholar] [CrossRef]
- Wu, W.; Qian, L.; Dai, J.; Ding, B.; Chen, X. Expression of chemokine CXCL12 and its receptor (CXCR4 and CXCR7) in different molecular subtypes of human breast carcinoma and the clinical significance. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2017, 42, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and p38 Protein Kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef]
- Rattanasinchai, C.; Gallo, K. MLK3 Signaling in Cancer Invasion. Cancers 2016, 8, 51. [Google Scholar] [CrossRef]
- Cronan, M.R.; Nakamura, K.; Johnson, N.L.; Granger, D.A.; Cuevas, B.D.; Wang, J.-G.; Mackman, N.; Scott, J.E.; Dohlman, H.G.; Johnson, G.L. Defining MAP3 kinases required for MDA-MB-231 cell tumor growth and metastasis. Oncogene 2012, 31, 3889–3900. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Xie, H.; Dou, Y.; Yuan, J.; Zeng, D.; Xiao, S. Expression and function of FRA1 protein in tumors. Mol. Biol. Rep. 2020, 47, 737–752. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Rattanasinchai, C.; Llewellyn, B.J.; Conrad, S.E.; Gallo, K.A. MLK3 regulates FRA-1 and MMPs to drive invasion and transendothelial migration in triple-negative breast cancer cells. Oncogenesis 2017, 6, e345. [Google Scholar] [CrossRef]
- Kohli, S.; Kohli, V. Role of RANKL-RANK/osteoprotegerin molecular complex in bone remodeling and its immunopathologic implications. Indian J. Endocr. Metab. 2011, 15, 175. [Google Scholar] [CrossRef] [PubMed]
- Cody, J.J.; Rivera, A.A.; Lyons, G.R.; Yang, S.W.; Wang, M.; Sarver, D.B.; Wang, D.; Selander, K.S.; Kuo, H.-C.; Meleth, S.; et al. Arming a replicating adenovirus with osteoprotegerin reduces the tumor burden in a murine model of osteolytic bone metastases of breast cancer. Cancer Gene 2010, 17, 893–905. [Google Scholar] [CrossRef][Green Version]
- Sheridan, J.P. Control of TRAIL-Induced Apoptosis by a Family of Signaling and Decoy Receptors. Science 1997, 277, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Geerts, D.; Chopra, C.; Connelly, L. Osteoprotegerin: Relationship to Breast Cancer Risk and Prognosis. Front. Oncol. 2020, 10, 462. [Google Scholar] [CrossRef]
- Zhong, Z.; Niu, P.; Wang, M.; Huang, G.; Xu, S.; Sun, Y.; Xu, X.; Hou, Y.; Sun, X.; Yan, Y.; et al. Targeted disruption of sp7 and myostatin with CRISPR-Cas9 results in severe bone defects and more muscular cells in common carp. Sci. Rep. 2016, 6, 22953. [Google Scholar] [CrossRef]
- Kichev, A.; Eede, P.; Gressens, P.; Thornton, C.; Hagberg, H. Implicating Receptor Activator of NF-κB (RANK)/RANK Ligand Signalling in Microglial Responses to Toll-Like Receptor Stimuli. Dev. Neurosci. 2017, 39, 192–206. [Google Scholar] [CrossRef]
- Goswami, S.; Sharma-Walia, N. Crosstalk between osteoprotegerin (OPG), fatty acid synthase (FASN) and, cycloxygenase-2 (COX-2) in breast cancer: Implications in carcinogenesis. Oncotarget 2016, 7, 58953–58974. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Luo, Z.-Q. Post-translational regulation of ubiquitin signaling. J. Cell Biol. 2019, 218, 1776–1786. [Google Scholar] [CrossRef]
- Shearer, R.F.; Iconomou, M.; Watts, C.K.W.; Saunders, D.N. Functional Roles of the E3 Ubiquitin Ligase UBR5 in Cancer. Mol. Cancer Res. 2015, 13, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Song, M.; Li, X.; Tang, L.; Zhang, T.; Zhang, L.; Pan, Y.; Chouchane, L.; Ma, X. E3 Ubiquitin Ligase UBR5 Drives the Growth and Metastasis of Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 2090–2101. [Google Scholar] [CrossRef]
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of Breast and Ovarian Cancers among BRCA1 and BRCA2 Mutation Carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 2012, 21, 134–147. [Google Scholar] [CrossRef]
- Wilson, J.R.F.; Bateman, A.C.; Hanson, H.; An, Q.; Evans, G.; Rahman, N.; Jones, J.L.; Eccles, D.M. A novel HER2-positive breast cancer phenotype arising from germline TP53 mutations. J. Med. Genet. 2010, 47, 771–774. [Google Scholar] [CrossRef]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef]
- Ahmed, A.; Ashraf, D.; Bahaa, A.; El-Tayebi, H.; Adwan, H. Impact of CDK4 knock out using CRISPR/Cas9 gene editing technology on breast cancer progression. Eur. J. Cancer 2020, 138, S70–S71. [Google Scholar] [CrossRef]
- Xu, K.; Chen, G.; Li, X.; Wu, X.; Chang, Z.; Xu, J.; Zhu, Y.; Yin, P.; Liang, X.; Dong, L. MFN2 suppresses cancer progression through inhibition of mTORC2/Akt signaling. Sci. Rep. 2017, 7, 41718. [Google Scholar] [CrossRef] [PubMed]
- Mendes de Almeida, R.; Bandarra, S.; Clara Ribeiro, A.; Mascarenhas, P.; Bekman, E.; Barahona, I. Inactivation of APOBEC3G gene in breast cancer cells using the CRISPR/Cas9 system. Ann. Med. 2019, 51, 40. [Google Scholar] [CrossRef]
- Heidary Arash, E.; Shiban, A.; Song, S.; Attisano, L. MARK4 inhibits Hippo signaling to promote proliferation and migration of breast cancer cells. EMBO Rep. 2017, 18, 420–436. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Fernández, M.; Sanz-Flores, M.; Sanz-Castillo, B.; Salazar-Roa, M.; Partida, D.; Zapatero-Solana, E.; Ali, H.R.; Manchado, E.; Lowe, S.; VanArsdale, T.; et al. Therapeutic relevance of the PP2A-B55 inhibitory kinase MASTL/Greatwall in breast cancer. Cell Death Differ. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Giuliano, C.J.; Sayles, N.M.; Sheltzer, J.M. CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on-going clinical trials. eLife 2017, 6, e24179. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wang, V.; Wang, N.; Khadang, B.; Boudreault, J.; Bakdounes, K.; Ali, S.; Lebrun, J.-J. Identification of MFGE8 and KLK5/7 as mediators of breast tumorigenesis and resistance to COX-2 inhibition. Breast Cancer Res. 2021, 23, 23. [Google Scholar] [CrossRef]
- Kim, D.; Bae, S.; Park, J.; Kim, E.; Kim, S.; Yu, H.R.; Hwang, J.; Kim, J.-I.; Kim, J.-S. Digenome-seq: Genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 2015, 12, 237–243. [Google Scholar] [CrossRef]
- Wilbie, D.; Walther, J.; Mastrobattista, E. Delivery Aspects of CRISPR/Cas for in Vivo Genome Editing. Acc. Chem. Res. 2019, 52, 1555–1564. [Google Scholar] [CrossRef]
- Kouranova, E.; Forbes, K.; Zhao, G.; Warren, J.; Bartels, A.; Wu, Y.; Cui, X. CRISPRs for Optimal Targeting: Delivery of CRISPR Components as DNA, RNA, and Protein into Cultured Cells and Single-Cell Embryos. Hum. Gene Ther. 2016, 27, 464–475. [Google Scholar] [CrossRef]
- Luther, D.C.; Lee, Y.W.; Nagaraj, H.; Scaletti, F.; Rotello, V.M. Delivery approaches for CRISPR/Cas9 therapeutics in vivo: Advances and challenges. Expert Opin. Drug Deliv. 2018, 15, 905–913. [Google Scholar] [CrossRef]
- Gray, S.J.; Woodard, K.T.; Samulski, R.J. Viral vectors and delivery strategies for CNS gene therapy. Ther. Deliv. 2010, 1, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, A.P.; Song, M.; Kim, K.-S.; Ramakrishna, S. Different Methods of Delivering CRISPR/Cas9 Into Cells. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2018; Volume 159, pp. 157–176. ISBN 978-0-12-816235-4. [Google Scholar]
- Rauschhuber, C.; Noske, N.; Ehrhardt, A. New insights into stability of recombinant adenovirus vector genomes in mammalian cells. Eur. J. Cell Biol. 2012, 91, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Crystal, R.G. Adenovirus: The First Effective In Vivo Gene Delivery Vector. Hum. Gene Ther. 2014, 25, 3–11. [Google Scholar] [CrossRef]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zeng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef]
- Xu, C.L.; Ruan, M.Z.C.; Mahajan, V.B.; Tsang, S.H. Viral Delivery Systems for CRISPR. Viruses 2019, 11, 28. [Google Scholar] [CrossRef]
- Ahi, Y.S.; Bangari, D.S.; Mittal, S.K. Adenoviral Vector Immunity: Its Implications and Circumvention Strategies. Curr. Gene Ther. 2011, 11, 307–320. [Google Scholar] [CrossRef]
- Ricobaraza, A.; Gonzalez-Aparicio, M.; Mora-Jimenez, L.; Lumbreras, S.; Hernandez-Alcoceba, R. High-Capacity Adenoviral Vectors: Expanding the Scope of Gene Therapy. Int. J. Mol. Sci. 2020, 21, 3643. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control Release 2017, 266, 17–26. [Google Scholar] [CrossRef]
- Wanisch, K.; Yáñez-Muñoz, R.J. Integration-deficient Lentiviral Vectors: A Slow Coming of Age. Mol. Ther. 2009, 17, 1316–1332. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Therapeutic in vivo gene transfer for genetic disease using AAV: Progress and challenges. Nat. Rev. Genet. 2011, 12, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.-H.; Suh, Y. In vivo genome editing in animals using AAV-CRISPR system: Applications to translational research of human disease. F1000Research 2017, 6, 2153. [Google Scholar] [CrossRef]
- Linden, R.M.; Ward, P.; Giraud, C.; Winocour, E.; Berns, K.I. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA 1996, 93, 11288–11294. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Gersbach, C.A. Engineering Delivery Vehicles for Genome Editing. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 637–662. [Google Scholar] [CrossRef] [PubMed]
- Hermonat, P.L.; Quirk, J.G.; Bishop, B.M.; Han, L. The packaging capacity of adeno-associated virus (AAV) and the potential for wild-type-plus AAV gene therapy vectors. FEBS Lett. 1997, 407, 78–84. [Google Scholar] [CrossRef]
- Ramamoorth, M. Non Viral Vectors in Gene Therapy—An Overview. J. Clin. Diagn. Res. 2015. [Google Scholar] [CrossRef]
- Young, J.L.; Dean, D.A. Electroporation-Mediated Gene Delivery. In Advances in Genetics; Elsevier: Amsterdam, The Netherlands, 2015; Volume 89, pp. 49–88. ISBN 978-0-12-802272-6. [Google Scholar]
- Rubinsky, B. Irreversible Electroporation in Medicine. Technol Cancer Res. Treat. 2007, 6, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Dean, D.A.; Gasiorowski, J.Z. Microinjecting Cells Using a Constant-Flow Microinjection System. Cold Spring Harb. Protoc. 2011, 2011, prot5590. [Google Scholar] [CrossRef][Green Version]
- Luong, L.N.; McFalls, K.M.; Kohn, D.H. Gene delivery via DNA incorporation within a biomimetic apatite coating. Biomaterials 2009, 30, 6996–7004. [Google Scholar] [CrossRef]
- Chowdhury, E.H.; Sasagawa, T.; Nagaoka, M.; Kundu, A.K.; Akaike, T. Transfecting mammalian cells by DNA/calcium phosphate precipitates: Effect of temperature and pH on precipitation. Anal. Biochem. 2003, 314, 316–318. [Google Scholar] [CrossRef]
- Daraee, H.; Etemadi, A.; Kouhi, M.; Alimirzalu, S.; Akbarzadeh, A. Application of liposomes in medicine and drug delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 381–391. [Google Scholar] [CrossRef]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 975. [Google Scholar] [CrossRef]
- Shalaby, K.; Aouida, M.; El-Agnaf, O. Tissue-Specific Delivery of CRISPR Therapeutics: Strategies and Mechanisms of Non-Viral Vectors. Int. J. Mol. Sci. 2020, 21, 7353. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR—Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, F.; Chen, Y.; Liu, J.; Wang, X.; Chen, A.T.; Deng, G.; Zhang, H.; Liu, J.; Hong, Z.; et al. Targeted Delivery of CRISPR/Cas9-Mediated Cancer Gene Therapy via Liposome-Templated Hydrogel Nanoparticles. Adv. Funct. Mater. 2017, 27, 1703036. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Cheng, Q.; Min, Y.-L.; Olson, E.N.; Siegwart, D.J. Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun. 2020, 11, 3232. [Google Scholar] [CrossRef] [PubMed]
- Maffei, M.; Morelli, C.; Graham, E.; Patriarca, S.; Donzelli, L.; Doleschall, B.; de Castro Reis, F.; Nocchi, L.; Chadick, C.H.; Reymond, L.; et al. A ligand-based system for receptor-specific delivery of proteins. Sci. Rep. 2019, 9, 19214. [Google Scholar] [CrossRef]
- Zhuang, J.; Tan, J.; Wu, C.; Zhang, J.; Liu, T.; Fan, C.; Li, J.; Zhang, Y. Extracellular vesicles engineered with valency-controlled DNA nanostructures deliver CRISPR/Cas9 system for gene therapy. Nucleic Acids Res. 2020, 48, 8870–8882. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef]
- Ding, Q.; Regan, S.N.; Xia, Y.; Oostrom, L.A.; Cowan, C.A.; Musunuru, K. Enhanced Efficiency of Human Pluripotent Stem Cell Genome Editing through Replacing TALENs with CRISPRs. Cell Stem Cell 2013, 12, 393–394. [Google Scholar] [CrossRef]
- Li, S.; Garay, J.P.; Tubbs, C.A.; Franco, H.L. CRISPR-based knock-in mutagenesis of the pioneer transcription factor FOXA1: Optimization of strategies for multi-allelic proteins in cancer cells. FEBS Open Bio. 2021. [Google Scholar] [CrossRef]
- Wang, W.; Malyutina, A.; Pessia, A.; Saarela, J.; Heckman, C.A.; Tang, J. Combined gene essentiality scoring improves the prediction of cancer dependency maps. EBioMedicine 2019, 50, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Wen, S.; Liu, F.; Zhang, Z.; Liu, M.; Wu, Y.; He, B.; Yan, M.; Kang, T.; Lam, E.W.; et al. CRISPR/Cas9 screening identifies a kinetochore-microtubule dependent mechanism for Aurora-A inhibitor resistance in breast cancer. Cancer Commun. 2021, 41, 121–139. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Hiranniramol, K.; Chen, Y.; Liu, W.; Wang, X. Generalizable sgRNA design for improved CRISPR/Cas9 editing efficiency. Bioinformatics 2020, 36, 2684–2689. [Google Scholar] [CrossRef]
- Walton, R.T.; Christie, K.A.; Whittaker, M.N.; Kleinstiver, B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 2020, 368, 290–296. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef]
- Baylis, F.; McLeod, M. First-in-human Phase 1 CRISPR Gene Editing Cancer Trials: Are We Ready? Curr. Gene Ther. 2018, 17. [Google Scholar] [CrossRef]
- Yu, K.-R.; Natanson, H.; Dunbar, C.E. Gene Editing of Human Hematopoietic Stem and Progenitor Cells: Promise and Potential Hurdles. Hum. Gene Ther. 2016, 27, 729–740. [Google Scholar] [CrossRef]
- Tong, S.; Moyo, B.; Lee, C.M.; Leong, K.; Bao, G. Engineered materials for in vivo delivery of genome-editing machinery. Nat. Rev. Mater. 2019, 4, 726–737. [Google Scholar] [CrossRef]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. The CRISPR-baby scandal: What’s next for human gene-editing. Nature 2019, 566, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Brokowski, C.; Adli, M. CRISPR Ethics: Moral Considerations for Applications of a Powerful Tool. J. Mol. Biol. 2019, 431, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Andorno, R.; Baylis, F.; Darnovsky, M.; Dickenson, D.; Haker, H.; Hasson, K.; Lowthorp, L.; Annas, G.J.; Bourgain, C.; Drabiak, K.; et al. Geneva Statement on Heritable Human Genome Editing: The Need for Course Correction. Trends Biotechnol. 2020, 38, 351–354. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CRIPSR/Cas Systems | ||||||
|---|---|---|---|---|---|---|
| Class | 1 | 2 | ||||
| Protein type | Multiplex | Single | ||||
| Type | I | III | IV | II | V | VI |
| Corresponding Cas protein | Cas 3 | Cas 10 | Cas 8 | Cas 9 | Cas 12a, Cas 12c, Cas 13a | Cas 13b, Cas 13c |
| Gene | Protein Encoded | Role in Breast Cancer | Associated Subtype of Breast Cancer | CRISPR/Cas9 Application | Results | Ref. |
|---|---|---|---|---|---|---|
| BRCA 1 | BRCA1 | Lacks DSB repair ability | TNBC | Nucleotide substitution through BE | Successful identification of LOF variants | [41,47,129] |
| BRCA 2 | BRCA2 | Lacks DSB repair ability | ER+ and HER2- | KO | Cell inviability | [51,53,129] |
| HER2 | HER2 | Promotes cell proliferation | HER2+ | KO | Suppressed cell proliferation and tumorigenesis | [55] |
| TP53 | p-53 | Deregulates cell cycle | TNBC and/or HER2+ | Cas9 expressed in cell lines | Cas9 induces TP53 mutation | [62,63,130] |
| TP53BP1 | 53BP1 | Lacks DSB repair ability | TNBC | KO | Restoration of HR in BRCA1 and P53BP1 deficient cells | [64,131] |
| MKI67 | Ki-67 | Promotes initiation, progression, metastasis | N/A | KO | Wide-spread transcriptome changes | [71] |
| MIEN1 | MIEN1 protein | Promotes initiation, metastasis | TNBC | KO | No effect on cell viability and tumor proliferation | [92] |
| CX3CR1 | CX3CR1 | Promotes skeletal and soft tissue organ metastasis | Luminal A&B, HER2+, TNBC | Transcription silencing | Reduction in skeletal and lung metastasis | [93] |
| CXCR2 | CXCR2 | Promotes cell proliferation, migration, angiogenesis | TNBC | KO | Suppressed cell proliferation, migration and metastasis | [104] |
| CXCR4 and CXCR7 | CXCR4 and CXCR7 | Promotes cell proliferation, invasion, metastasis | TNBC | Single-gene knockout and co-knockout | Suppressed cell proliferation, invasion and migration | [106] |
| MAP3K11 | MLK 3 | Promotes metastasis | TNBC | KO | Suppressed cell invasion and migration | [118] |
| TNFRSF11B | OPG | Blocks apoptosis | HR+ | KO | Inhibited protein expression of Fatty Acid Synthase | [125] |
| UBR5 | Ubr5 | Aids EMT | TNBC | KO | Induced apoptosis and suppressed metastasis | [128] |
| CDK4 | CDK4 | Cell proliferation | TNBC and HER2+ | KO | Suppressed viability, clono-genicity, migration | [132] |
| MFN2 | MFN2 | Suppress cancer progression via mTOR2/Akt signal inhibition | HR+ | KO | Promotes cell viability, invasion, colony formation | [133] |
| APOBEC3G | APOBEC3 | APOBEC3 induced mutagenesis | HER2+ | KO | Suppressed cell proliferation | [134] |
| MARK4 | MARK4 | Inhibits Hippo signaling leading to cell proliferation | TNBC | KO | Suppressed cell proliferation and migration | [135] |
| MASTL | MASTL | Promotes proliferation | HR+ and TNBC | KO | Suppressed proliferation and tumor growth | [136] |
| MELK | MELK | Tumorigenesis regulator | TNBC | KO | CRISPR/Cas9-Mediated Mutagenesis | [137] |
| MFGE8 | MFGE8 | Mediator of breast cancer tumorigenesis | TNBC | KO | Restored sensitivity to COX-2 selective inhibitor | [138] |
| KLK5 | KLK5 | Serum biomarker | TNBC | KO | ||
| KLK7 | KLK7 |
| Vector | Mechanism | Immunogenicity | Packaging Capacity (kb) | Reference |
|---|---|---|---|---|
| Lentivirus | Incorporates into host genome | ++ | 9.7 | [144] |
| Adenovirus | Do not integrate; remain extrachromosomal | +++ | 35 | [148] |
| Adeno-mediated adenovirus | May incorporate; primarily remain extrachromosomal | + | 4.7 | [142,157] |
| Method | Mechanism | Advantage | Disadvantage | Reference |
|---|---|---|---|---|
| Electroporation | Creates pores in plasma membrane; increases permeability | No size restrictions | High cellular lysis | [158] |
| Microinjection | Ejection of genetic material using a micro-pipette | Good reproducibility | Lower throughput | [151] |
| Calcium Phosphonate | Blending genetic material with CaPO4 improves cellular adhesion and entry | Affordable | High cytotoxicity | [163] |
| Lipid-mediated | Liposomal fusion with cell membrane to facilitate access into cell | High transfection rates | Culture condition affect efficiency | [165] |
| Cancer | Phase | Intervention | Country | ID |
|---|---|---|---|---|
| Gastrointestinal, Epithelial Cancer, Gastrointestinal Neoplasms, Cancer of Gastrointestinal Tract, Cancer, Gastrointestinal, Gastrointestinal Cancer, Colo-rectal Cancer, Pancreatic Cancer, Gall Bladder Cancer, Colon Cancer, Esophageal Cancer, Stomach Cancer | I/II | Drug: Cyclophosphamide Drug: Fludarabine Biological: Tumor-Infiltrating Lymphocytes (TIL) Drug: Aldesleukin | USA | NCT04426669 |
| Solid Tumor, Adult | I | Biological: anti-mesothelin CAR-T cells | China | NCT03545815 |
| B-cell Malignancy, Non-Hodgkin Lymphoma, B-cell Lymphoma, Adult B Cell ALL | I | Biological: CTX110 | USA | NCT04035434 |
| T Cell Lymphoma | I | Biological: CTX130 | USA | NCT04502446 |
| Multiple Myeloma | I | Biological: CTX120 | USA | NCT04244656 |
| B Cell Leukemia, B Cell Lymphoma | I/II | Biological: UCART019 | China | NCT03166878 |
| Renal Cell Carcinoma | I | Biological: CTX130 | USA | NCT04438083 |
| B Acute Lymphoblastic Leukemia | I | Drug: PBLTT52CAR19 | UK | NCT04557436 |
| B Cell Leukemia, B Cell Lymphoma | I/II | Biological: Universal Dual Specificity CD19 and CD20 or CD22 CAR-T Cells | China | NCT03398967 |
| Gene Studied | Prospective Application |
|---|---|
| BRCA1 | Reclassification of BRCA1 and potentially other breast cancer VUSs through CRISPR-based high-throughput screens. |
| BRCA2 | Further studies are needed to understand how knock-out of BRCA2, a tumor suppressor, leads to cell inviability, yet this is overcome in BRCA2 mutated breast cancers. |
| HER2 | CRISPR/Cas9 can be used to target HER2 to achieve a therapeutic outcome in clinical settings. |
| TP53 | The study highlighted a limitation when conducting future experiments on TP53 with Cas9. |
| TP53BP1 | PALB2 could be tested for its clinical implications in clinically significant BRCA1/53BP1 deficient cells. |
| MKI67 | Further studies on identifying the interacting proteins and effectors involved in MKI67 knock-out can help identify therapeutic targets. |
| CXCR4 and CXCR7 | These genes can function as possible therapeutic targets for TNBC treatment. |
| MAP3K11 | This gene can function as a possible therapeutic target for TNBC treatment. |
| CX3CR1 | Can be targeted to limit breast cancer metastasis to the bone. |
| CXCR2 | This gene can function as a possible therapeutic target for TNBC treatment. |
| UBR5 | This gene can function as a possible therapeutic target for TNBC treatment. |
| MIEN1 | Further studies are required on this gene to substantiate the function of MIEN1 in metastasis. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, M.; Daoud, G.H.; Mohamed, A.; Harati, R. New Insights into the Therapeutic Applications of CRISPR/Cas9 Genome Editing in Breast Cancer. Genes 2021, 12, 723. https://doi.org/10.3390/genes12050723
Ahmed M, Daoud GH, Mohamed A, Harati R. New Insights into the Therapeutic Applications of CRISPR/Cas9 Genome Editing in Breast Cancer. Genes. 2021; 12(5):723. https://doi.org/10.3390/genes12050723
Chicago/Turabian StyleAhmed, Munazza, Grace Hope Daoud, Asmaa Mohamed, and Rania Harati. 2021. "New Insights into the Therapeutic Applications of CRISPR/Cas9 Genome Editing in Breast Cancer" Genes 12, no. 5: 723. https://doi.org/10.3390/genes12050723
APA StyleAhmed, M., Daoud, G. H., Mohamed, A., & Harati, R. (2021). New Insights into the Therapeutic Applications of CRISPR/Cas9 Genome Editing in Breast Cancer. Genes, 12(5), 723. https://doi.org/10.3390/genes12050723