
Adaptive Divergence under Gene Flow along an Environmental Gradient in Two Coexisting Stickleback Species

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
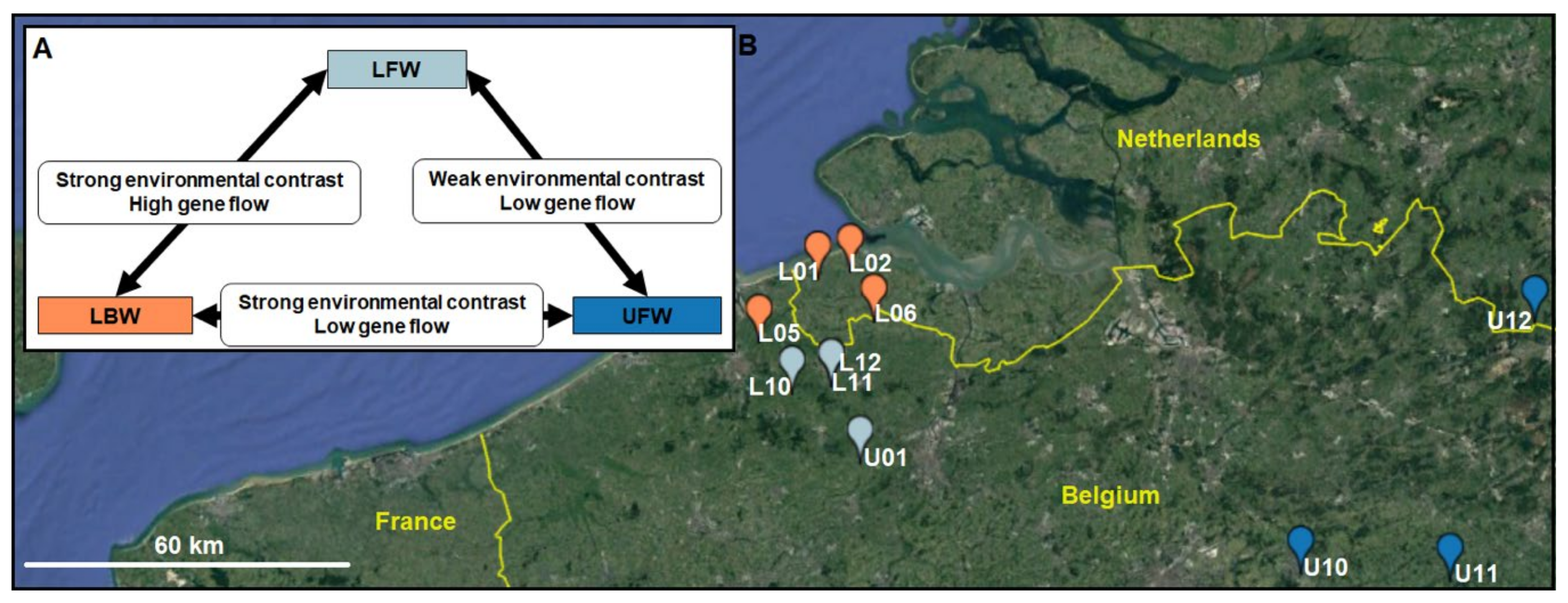
2.1. Study Area and Sampling Design
2.2. Morphological Characterisation
2.3. DNA Extraction, Genotyping-by-Sequencing and SNP Filtering
2.4. Population Structure and Genetic Diversity Statistics
2.5. Signatures of Adaptive Divergence
3. Results
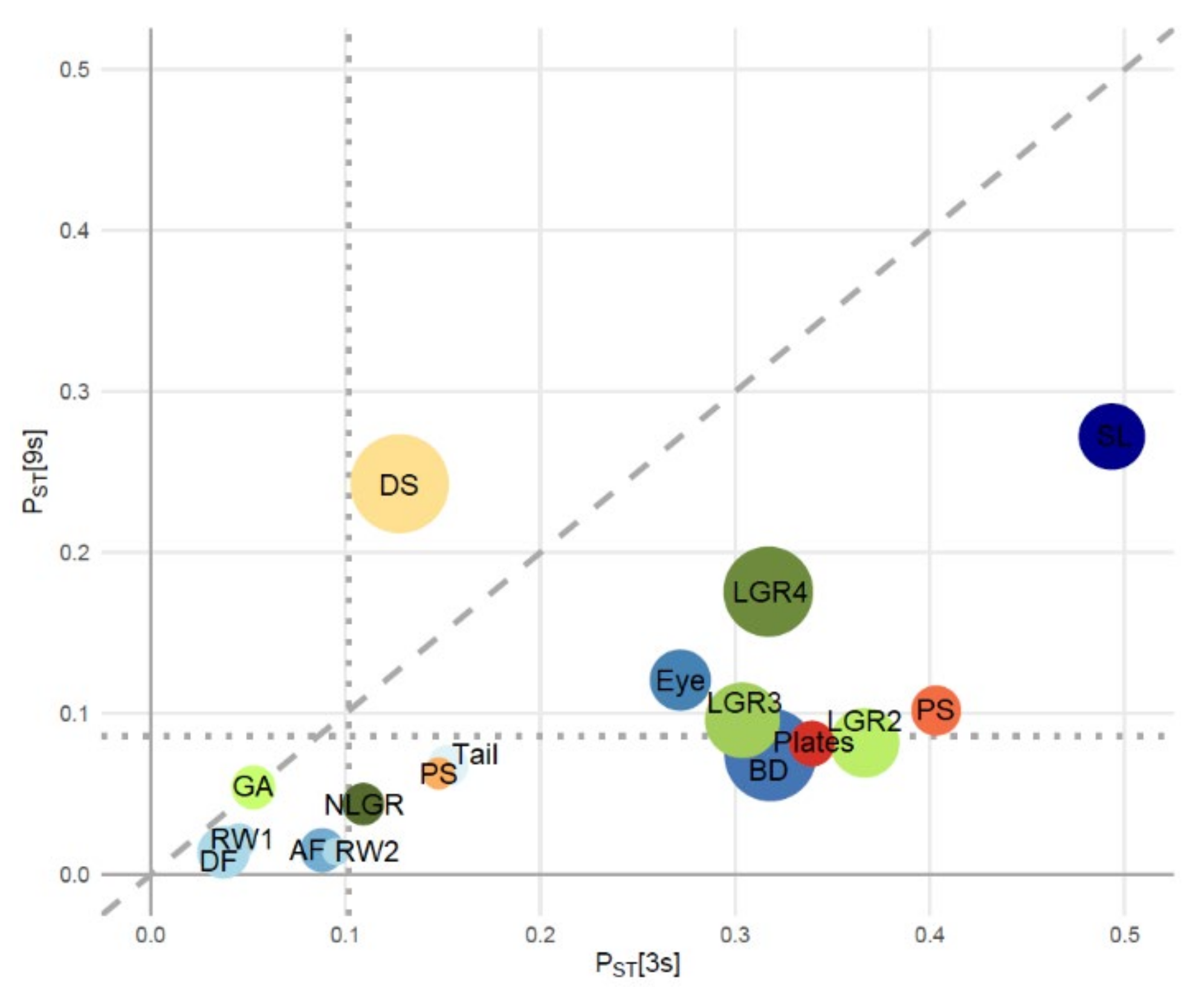
3.1. Morphological Divergence
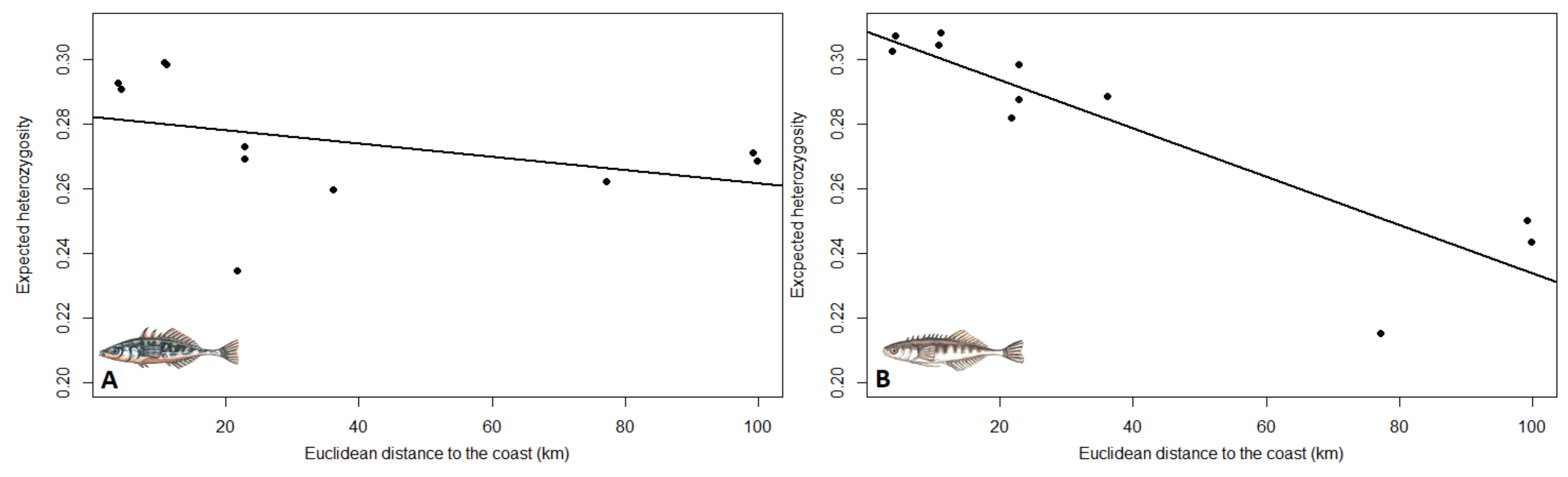
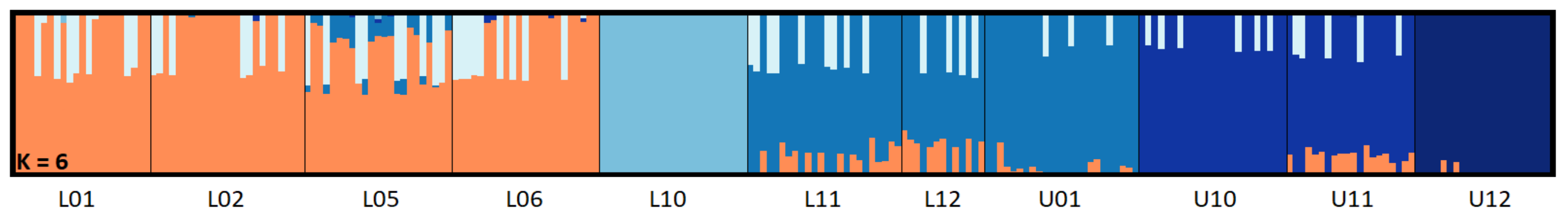
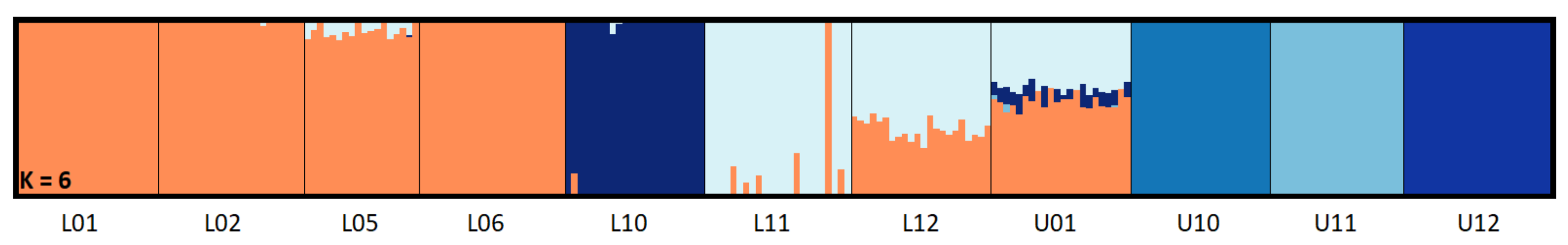
3.2. Population Structure and Genetic Diversity Statistics
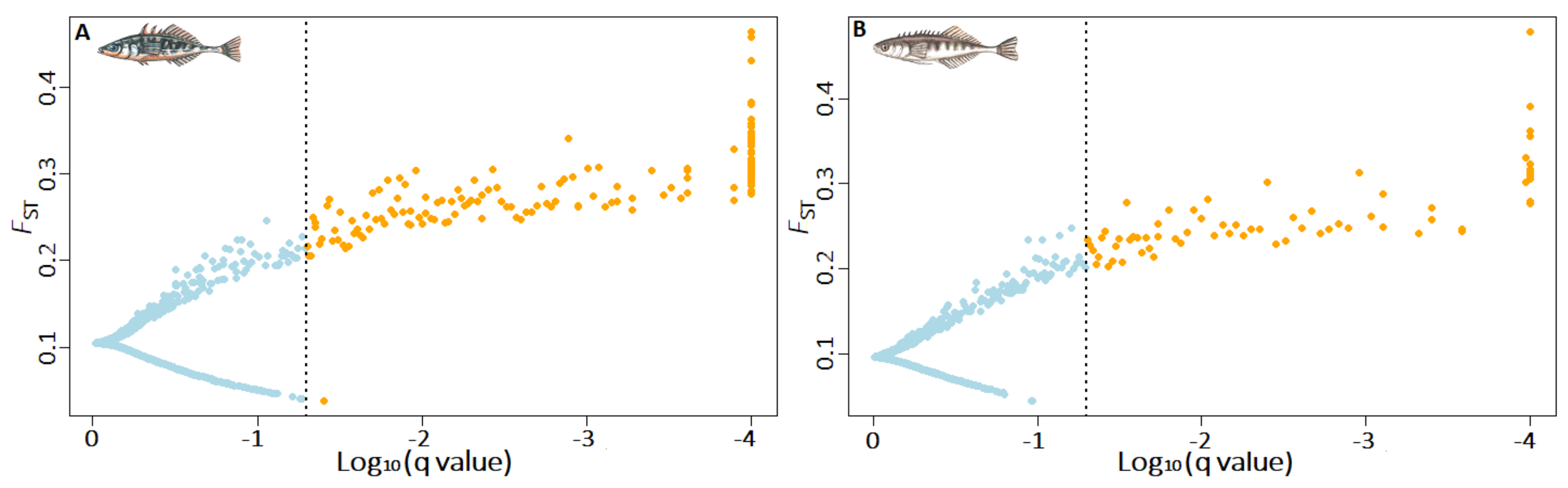
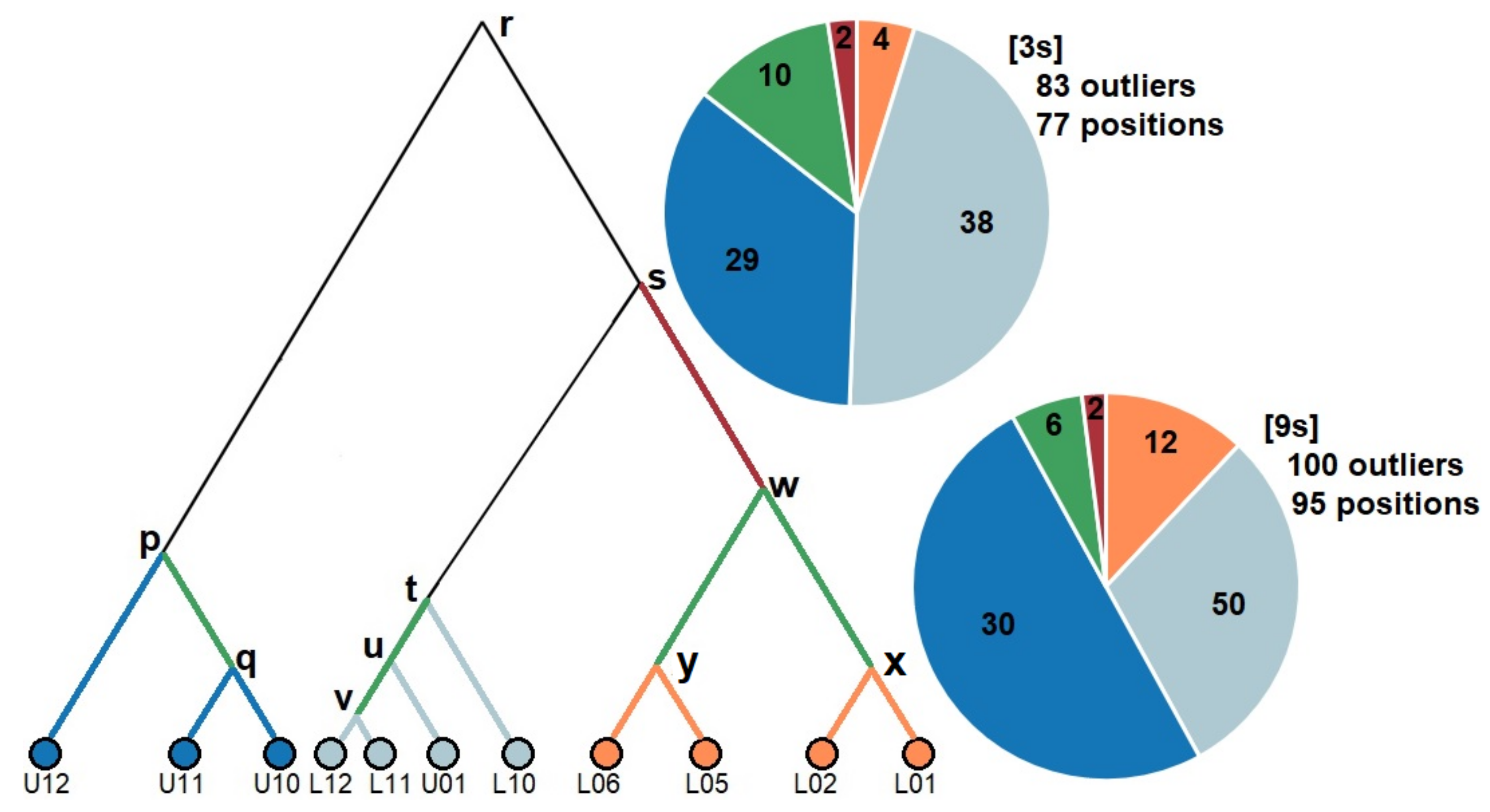
3.3. Signatures of Adaptive Divergence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levins, R. The theory of fitness in a heterogeneous environment. IV. The Adaptive Significance of Gene Flow. Evolution 1964, 18, 635. [Google Scholar] [CrossRef]
- Endler, J.A. Gene flow and population differentiation: Studies of clines suggest that differentiation along environmental gradients may be independent of gene flow. Science 1973, 179, 243–250. [Google Scholar] [CrossRef]
- Tigano, A.; Friesen, V.L. Genomics of local adaptation with gene flow. Mol. Ecol. 2016, 25, 2144–2164. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.P.; Forester, B.R.; Latch, E.K.; Aitken, S.N.; Hoban, S. Guidelines for planning genomic assessment and monitoring of locally adaptive variation to inform species conservation. Evol. Appl. 2018, 11, 1035–1052. [Google Scholar] [CrossRef] [PubMed]
- Hällfors, M.H.; Liao, J.; Dzurisin, J.; Grundel, R.; Hyvärinen, M.; Towle, K.; Wu, G.C.; Hellmann, J.J. Addressing potential local adaptation in species distribution models: Implications for conservation under climate change. Ecol. Appl. 2016, 26, 1154–1169. [Google Scholar] [CrossRef]
- Kelly, E.; Phillips, B.L. Targeted gene flow and rapid adaptation in an endangered marsupial. Conserv. Biol. 2019, 33, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Phillips, B.L. Targeted gene flow for conservation. Conserv. Biol. 2016, 30, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Bernatchez, L. On the maintenance of genetic variation and adaptation to environmental change: Considerations from population genomics in fishes. J. Fish Biol. 2016, 89, 2519–2556. [Google Scholar] [CrossRef]
- Pfeifer, S.P.; Laurent, S.; Sousa, V.C.; Linnen, C.R.; Foll, M.; Excoffier, L.; Hoekstra, H.E.; Jensen, J.D. The evolutionary history of Nebraska deer mice: Local adaptation in the face of strong gene flow. Mol. Biol. Evol. 2018, 35, 792–806. [Google Scholar] [CrossRef]
- Diopere, E.; Vandamme, S.G.; Hablützel, P.I.; Cariani, A.; Van Houdt, J.; Rijnsdorp, A.; Tinti, F.; Volckaert, F.A.M.; Maes, G.E. FishPopTrace Consortium Seascape genetics of a flatfish reveals local selection under high levels of gene flow. ICES J. Mar. Sci. 2018, 75, 675–689. [Google Scholar] [CrossRef]
- Moody, K.N.; Hunter, S.N.; Childress, M.J.; Blob, R.W.; Schoenfuss, H.L.; Blum, M.J.; Ptacek, M.B. Local adaptation despite high gene flow in the waterfall-climbing Hawaiian goby. Sicyopterus Stimpsoni. Mol. Ecol. 2015, 24, 545–563. [Google Scholar] [CrossRef] [PubMed]
- Hablützel, P.I.; Grégoir, A.F.; Vanhove, M.P.M.; Volckaert, F.A.M.; Raeymaekers, J.A.M. Weak link between dispersal and parasite community differentiation or immunogenetic divergence in two sympatric cichlid fishes. Mol. Ecol. 2016, 25, 5451–5466. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.-S.; Hendry, A.P. Both selection and gene flow are necessary to explain adaptive divergence: Evidence from clinal variation in stream stickleback. Evol. Ecol. Res. 2005, 7, 871–886. [Google Scholar]
- Bachmann, J.C.; Jansen van Rensburg, A.; Cortazar-Chinarro, M.; Laurila, A.; Van Buskirk, J. Gene flow limits adaptation along steep environmental gradients. Am. Nat. 2020, 195, E67–E86. [Google Scholar] [CrossRef]
- Cornwell, B.H. Gene flow in the anemone Anthopleura elegantissima limits signatures of local adaptation across an extensive geographic range. Mol. Ecol. 2020, 29, 2550–2566. [Google Scholar] [CrossRef] [PubMed]
- Kalske, A.; Leimu, R.; Scheepens, J.F.; Mutikainen, P. Spatiotemporal variation in local adaptation of a specialist insect herbivore to its long-lived host plant. Evolution 2016, 70, 2110–2122. [Google Scholar] [CrossRef] [PubMed]
- Dennenmoser, S.; Vamosi, S.M.; Nolte, A.W.; Rogers, S.M. Adaptive genomic divergence under high gene flow between freshwater and brackish-water ecotypes of prickly sculpin (Cottus asper) revealed by Pool-Seq. Mol. Ecol. 2017, 26, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Pinho, C.; Hey, J. Divergence with gene Flow: Nodels and data. Annu. Rev. Ecol. Evol. Syst. 2010, 41, 215–230. [Google Scholar] [CrossRef]
- Feder, J.L.; Egan, S.P.; Nosil, P. The genomics of speciation-with-gene-flow. Trends Genet. 2012, 28, 342–350. [Google Scholar] [CrossRef]
- Oke, K.B.; Rolshausen, G.; LeBlond, C.; Hendry, A.P. How parallel is parallel evolution? A comparative analysis in fishes. Am. Nat. 2017, 190, 1–16. [Google Scholar] [CrossRef]
- Ferchaud, A.-L.; Hansen, M.M. The impact of selection, gene flow and demographic history on heterogeneous genomic divergence: Three-spine sticklebacks in divergent environments. Mol. Ecol. 2016, 25, 238–259. [Google Scholar] [CrossRef]
- Raeymaekers, J.A.M.; Chaturvedi, A.; Hablützel, P.I.; Verdonck, I.; Hellemans, B.; Maes, G.E.; De Meester, L.; Volckaert, F.A.M. Adaptive and non-adaptive divergence in a common landscape. Nat. Commun. 2017, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- DeFaveri, J.; Shikano, T.; Ghani, N.I.A.; Merilä, J. Contrasting population structures in two sympatric fishes in the Baltic Sea basin. Mar. Biol. 2012, 159, 1659–1672. [Google Scholar] [CrossRef]
- Copp, G.H.; Kováč, V. Sympatry between threespine Gasterosteus aculeatus and ninespine Pungitius pungitius sticklebacks in English lowland streams. In Annales Zoologici Fennici; Finnish Zoological and Botanical Publishing Board: Helsinki, Finland, 2003; Volume 40, pp. 341–355. [Google Scholar]
- Raeymaekers, J.A.M.; Huyse, T.; Maelfait, H.; Hellemans, B.; Volckaert, F.A.M. Community structure, population structure and topographical specialisation of Gyrodactylus (monogenea) ectoparasites living on sympatric stickleback species. Folia Parasitol. 2008, 55, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Copp, G.H.; Edmonds-Brown, V.R.; Cottey, R. Behavioural interactions and microhabitat use of stream-dwelling sticklebacks Gasterosteus aculateus and Pungitius pungitius in the laboratory and field. Folia Zool. Praha 1998, 47, 275–286. [Google Scholar]
- Lenz, T.L.; Eizaguirre, C.; Kalbe, M.; Milinski, M. Evaluating patterns of convergent evolution and trans-species polymorphism at MHC immunogenes in two sympatric stickleback species: Mhc evolution in two sympatric stickleback species. Evolution 2013, 67, 2400–2412. [Google Scholar] [CrossRef]
- Guo, B.; Chain, F.J.J.; Bornberg-Bauer, E.; Leder, E.H.; Merilä, J. Genomic divergence between nine- and three-spined sticklebacks. BMC Genom. 2013, 14, 756. [Google Scholar] [CrossRef][Green Version]
- Varadharajan, S.; Rastas, P.; Löytynoja, A.; Matschiner, M.; Calboli, F.C.F.; Guo, B.; Nederbragt, A.J.; Jakobsen, K.S.; Merilä, J. A high-quality assembly of the nine-Spined stickleback (Pungitius pungitius) Genome. Genome Biol. Evol. 2019, 11, 3291–3308. [Google Scholar] [CrossRef]
- Fang, B.; Merilä, J.; Matschiner, M.; Momigliano, P. Estimating uncertainty in divergence times among three-spined stickleback clades using the multispecies coalescent. Mol. Phylogenet. Evol. 2020, 142, 106646. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, H.S.; Merilä, J. Mitochondrial DNA phylogeography of the three-spined stickleback (Gasterosteus aculeatus) in Europe—Evidence for multiple glacial refugia. Mol. Phylogenet. Evol. 2008, 46, 167–182. [Google Scholar] [CrossRef]
- Wang, C.; Shikano, T.; Persat, H.; Merilä, J. Mitochondrial phylogeography and cryptic divergence in the stickleback genus Pungitius. J. Biogeogr. 2015, 42, 2334–2348. [Google Scholar] [CrossRef]
- Raeymaekers, J.A.M.; Konijnendijk, N.; Larmuseau, M.H.D.; Hellemans, B.; De Meester, L.; Volckaert, F.A.M. A gene with major phenotypic effects as a target for selection vs. homogenizing gene flow. Mol. Ecol. 2014, 23, 162–181. [Google Scholar] [CrossRef]
- Rohlf, F.J. The tps series of software. In Hystrix; Academia: San Francisco, CA, USA, 2015; Volume 26. [Google Scholar]
- Sharpe, D.M.T.; Räsänen, K.; Berner, D.; Hendry, A.P. Genetic and environmental contributions to the morphology of lake and stream stickleback: Implications for gene flow and reproductive isolation. Evol. Ecol. Res. 2008, 10, 849–866. [Google Scholar]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef] [PubMed]
- Jones, F.C.; Grabherr, M.G.; Chan, Y.F.; Russell, P.; Mauceli, E.; Johnson, J.; Swofford, R.; Pirun, M.; Zody, M.C.; White, S.; et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 2012, 484, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Lischer, H.E.L.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 2012, 28, 298–299. [Google Scholar] [CrossRef]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef]
- Li, Y.L.; Liu, J.X. StructureSelector: A web-based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Resour. 2018, 18, 176–177. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R foundation for statistical computing: Vienna, Austria, 2013. [Google Scholar]
- Goudet, J. Hierfstat, a package for r to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27, 209–220. [Google Scholar] [PubMed]
- Leinonen, T.; Cano, J.M.; Mäkinen, H.; Merilä, J. Contrasting patterns of body shape and neutral genetic divergence in marine and lake populations of threespine sticklebacks. J. Evol. Biol. 2006, 19, 1803–1812. [Google Scholar] [CrossRef]
- Raeymaekers, J.A.M.; Van Houdt, J.K.J.; Larmuseau, M.H.D.; Geldof, S.; Volckaert, F.A.M. Divergent selection as revealed by PST and QTL-based PST in three-spined stickleback (Gasterosteus aculeatus) populations along a coastal-inland gradient. Mol. Ecol. 2007, 16, 891–905. [Google Scholar] [CrossRef]
- Lunn, D.J.; Thomas, A.; Best, N.; Spiegelhalter, D. WinBUGS – a Bayesian modelling framework: Concepts, structure and extensibility. Statistics Comput. 2000, 10, 325–337. [Google Scholar] [CrossRef]
- Foll, M.; Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics 2008, 180, 977–993. [Google Scholar] [CrossRef]
- Refoyo-Martínez, A.; da Fonseca, R.R.; Halldórsdóttir, K.; Árnason, E.; Mailund, T.; Racimo, F. Identifying loci under positive selection in complex population histories. Genome Res. 2019, 29, 1506–1520. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Wang, Y.; Li, Z.; Guo, B.; Merilä, J. Population transcriptomics reveals weak parallel genetic basis in repeated marine and freshwater divergence in nine-spined sticklebacks. Mol. Ecol. 2020, 29, 1642–1656. [Google Scholar] [CrossRef]
- Kemppainen, P.; Li, Z.; Rastas, P.; Löytynoja, A.; Fang, B.; Guo, B.; Shikano, T.; Yang, J.; Merilä, J. Genetic population structure constrains local adaptation and probability of parallel evolution in sticklebacks. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shapiro, M.D.; Summers, B.R.; Balabhadra, S.; Aldenhoven, J.T.; Miller, A.L.; Cunningham, C.B.; Bell, M.A.; Kingsley, D.M. The genetic architecture of skeletal convergence and sex determination in ninespine sticklebacks. Curr. Biol. 2009, 19, 1140–1145. [Google Scholar] [CrossRef]
- Yeaman, S.; Whitlock, M.C. The genetic architecture of adaptation under migration–selection balance. Evolution 2011, 656, 1897–1911. [Google Scholar] [CrossRef]
- Fang, B.; Kemppainen, P.; Momigliano, P.; Merilä, J. Population structure limits parallel evolution. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lowe, W.H.; Kovach, R.P.; Allendorf, F.W. Population Genetics and Demography Unite Ecology and Evolution. Trends Ecol. Evol. 2017, 32, 141–152. [Google Scholar] [CrossRef]
- Van der Molen, J.; De Swart, H.E. Holocene tidal conditions and tide-induced sand transport in the southern North Sea. J. Geophys. Res. C Oceans 2001, 106, 9339–9362. [Google Scholar] [CrossRef]
- Teacher, A.G.F.; Shikano, T.; Karjalainen, M.E.; Merilä, J. Phylogeography and genetic structuring of european nine-spined sticklebacks (Pungitius pungitius)—mitochondrial DNA evidence. PLoS ONE 2011, 6, e19476. [Google Scholar] [CrossRef]
- Bell, M.A.; Andrews, C.A. Evolutionary consequences of postglacial colonization of fresh water by primitively anadromous fishes. In Evolutionary Ecology of Freshwater Animals: Concepts and Case Studies; Streit, B., Städler, T., Lively, C.M., Eds.; Birkhäuser Basel: Basel, Switzerland, 1997; pp. 323–363. ISBN 9783034888806. [Google Scholar]
- Raeymaekers, J.A.M.; Maes, G.E.; Audenaert, E.; Volckaert, F.A.M. Detecting Holocene divergence in the anadromous--freshwater three-spined stickleback (Gasterosteus aculeatus) system. Mol. Ecol. 2005, 14, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Booker, T.R.; Yeaman, S.; Whitlock, M.C. Variation in recombination rate affects detection of outliers in genome scans under neutrality. Mol. Ecol. 2020, 29, 4274–4279. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.B.; Walkey, M.; Dartnall, H.J.G. Some effects of low oxygen tensions on the distribution of the three-spined stickleback Gasterosteus aculeatus L. and the nine-spined stickleback Pungitius pungitius (L.). J. Fish Biol. 1972, 4, 103–108. [Google Scholar] [CrossRef]
- Kovac, V.; Copp, G.H.; Dimart, Y.; Uzikova, M. Comparative morphology of threespine Gasterosteus aculeatus and ninespine Pungitius pungitius sticklebacks in lowland streams of southeastern England. Folia Zool. Praha 2002, 51, 319–336. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | DTC | n[3s] | Ho[3s] | He[3s] | FIS[3s] | Ne[3s] | n[9s] | Ho[9s] | He[9s] | FIS [9s] | Ne[9s] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| L01 | 3.94 | 21 | 0.266 | 0.292 | 0.0805 | 213.4 | 22 | 0.246 | 0.303 | 0.167 | 441.7 |
| L02 | 4.30 | 24 | 0.266 | 0.290 | 0.0769 | 166.4 | 23 | 0.299 | 0.307 | 0.032 | 429.7 |
| L05 | 10.90 | 23 | 0.282 | 0.299 | 0.0550 | 198.4 | 18 | 0.240 | 0.304 | 0.187 | 142.0 |
| L06 | 11.14 | 23 | 0.294 | 0.298 | 0.0186 | 166.5 | 23 | 0.299 | 0.309 | 0.031 | 185.0 |
| L10 | 21.75 | 23 | 0.229 | 0.234 | 0.0249 | 120.8 | 22 | 0.266 | 0.282 | 0.052 | 690.2 |
| L11 | 22.84 | 24 | 0.229 | 0.273 | 0.141 | 72.2 | 23 | 0.291 | 0.288 | 0.001 | 127.4 |
| L12 | 22.84 | 13 | 0.159 | 0.269 | 0.344 | 814.0 | 22 | 0.283 | 0.299 | 0.052 | 508.3 |
| U01 | 36.20 | 24 | 0.232 | 0.259 | 0.097 | 219.5 | 22 | 0.283 | 0.289 | 0.022 | 575.5 |
| U10 | 77.10 | 23 | 0.239 | 0.262 | 0.081 | 137.2 | 22 | 0.163 | 0.215 | 0.213 | 177.1 |
| U11 | 99.20 | 20 | 0.261 | 0.271 | 0.034 | 138.8 | 21 | 0.219 | 0.250 | 0.114 | 344.5 |
| U12 | 99.80 | 21 | 0.253 | 0.268 | 0.053 | 94.8 | 23 | 0.175 | 0.244 | 0.251 | 271.2 |
| Geographical Scale | FST [3s] | Mean PST [3s] | PST > FST [3s] | FST [9s] | Mean PST [9s] | PST > FST [9s] |
|---|---|---|---|---|---|---|
| LBW–LFW–UFW | 0.102 | 0.22 | 7/17 (41%) | 0.086 | 0.090 | 2/17 (12%) |
| LBW–LFW | 0.081 | 0.19 | 7/17 (41%) | 0.040 | 0.071 | 1/17 (6%) |
| LBW–UFW | 0.071 | 0.20 | 7/17 (41%) | 0.097 | 0.093 | 2/17 (12%) |
| LFW–UFW | 0.140 | 0.19 | 4/17 (23%) | 0.118 | 0.100 | 1/17(6%) |
| Geographical Scale | Total Outliers [3s] | Proportion [3s] | Total Outliers [9s] | Proportion [9s] |
|---|---|---|---|---|
| LBW–LFW–UFW | 142 | 1.31% | 70 | 0.46% |
| LBW–LFW | 98 | 0.90% | 44 | 0.29% |
| LBW–UFW | 87 | 0.80% | 61 | 0.41% |
| LFW–UFW | 83 | 0.77% | 31 | 0.21% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bal, T.M.P.; Llanos-Garrido, A.; Chaturvedi, A.; Verdonck, I.; Hellemans, B.; Raeymaekers, J.A.M. Adaptive Divergence under Gene Flow along an Environmental Gradient in Two Coexisting Stickleback Species. Genes 2021, 12, 435. https://doi.org/10.3390/genes12030435
Bal TMP, Llanos-Garrido A, Chaturvedi A, Verdonck I, Hellemans B, Raeymaekers JAM. Adaptive Divergence under Gene Flow along an Environmental Gradient in Two Coexisting Stickleback Species. Genes. 2021; 12(3):435. https://doi.org/10.3390/genes12030435
Chicago/Turabian StyleBal, Thijs M. P., Alejandro Llanos-Garrido, Anurag Chaturvedi, Io Verdonck, Bart Hellemans, and Joost A. M. Raeymaekers. 2021. "Adaptive Divergence under Gene Flow along an Environmental Gradient in Two Coexisting Stickleback Species" Genes 12, no. 3: 435. https://doi.org/10.3390/genes12030435
APA StyleBal, T. M. P., Llanos-Garrido, A., Chaturvedi, A., Verdonck, I., Hellemans, B., & Raeymaekers, J. A. M. (2021). Adaptive Divergence under Gene Flow along an Environmental Gradient in Two Coexisting Stickleback Species. Genes, 12(3), 435. https://doi.org/10.3390/genes12030435