
Filling the Gaps in the Cyanobacterial Tree of Life—Metagenome Analysis of Stigonema ocellatum DSM 106950, Chlorogloea purpurea SAG 13.99 and Gomphosphaeria aponina DSM 107014


Abstract
1. Introduction
2. Material and Methods
2.1. Strains and Cultivation Conditions
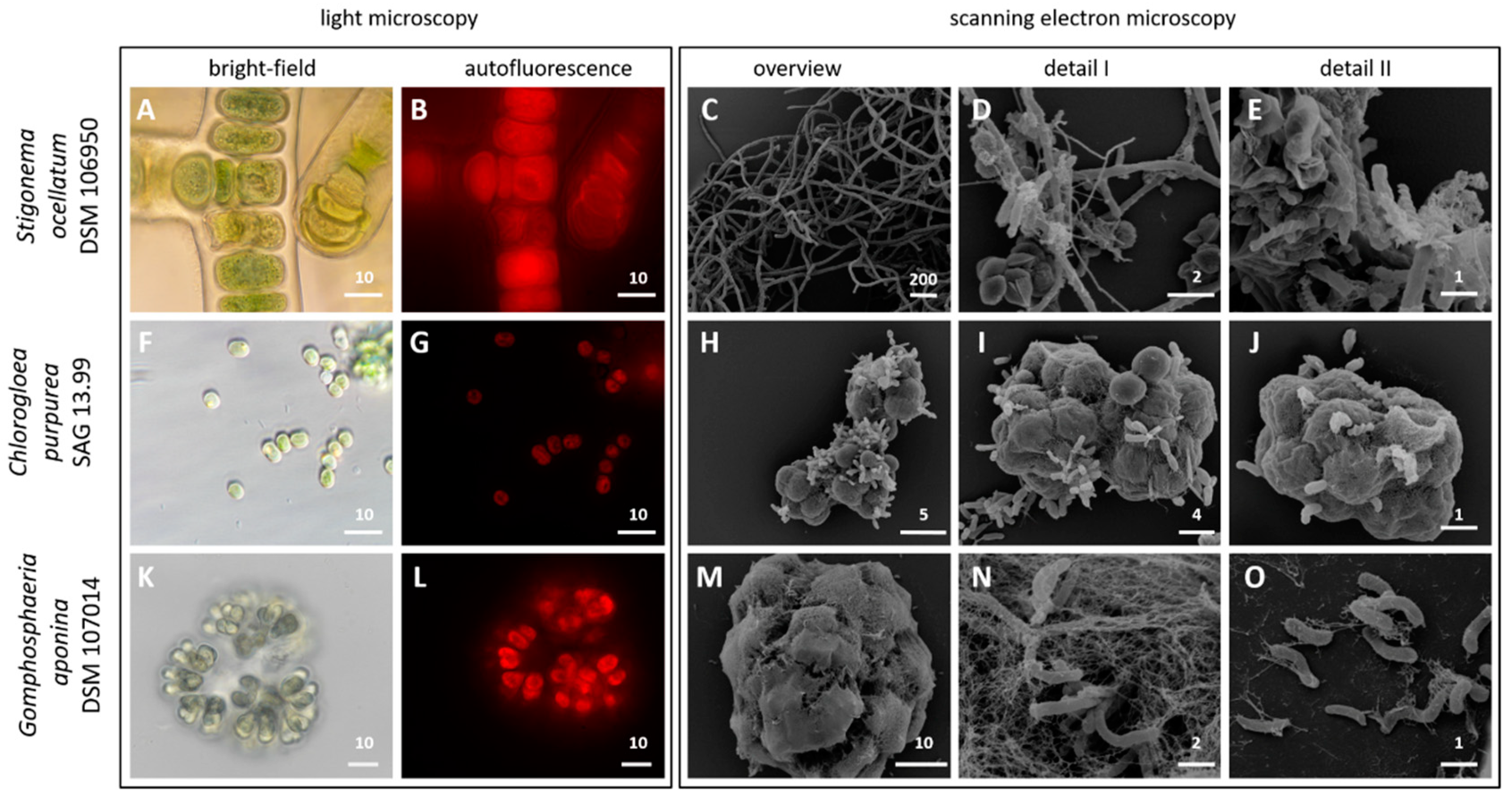
2.2. Light and Electron Microscopy
2.3. Metagenomics
2.3.1. Metagenome Sequencing
2.3.2. Metagenome Assembly and Binning
2.4. Taxonomic Assessment via BLASTN-Dependent Text Mining
2.5. Phylogenetic Analyses
2.6. Manual Curation of Metagenome Assembled Genomes (MAGs)
3. Results
3.1. Light and Scanning Electron Microscopy
3.2. Metagenome Sequencing of Three Non-Axenic Freshwater Cyanobacteria
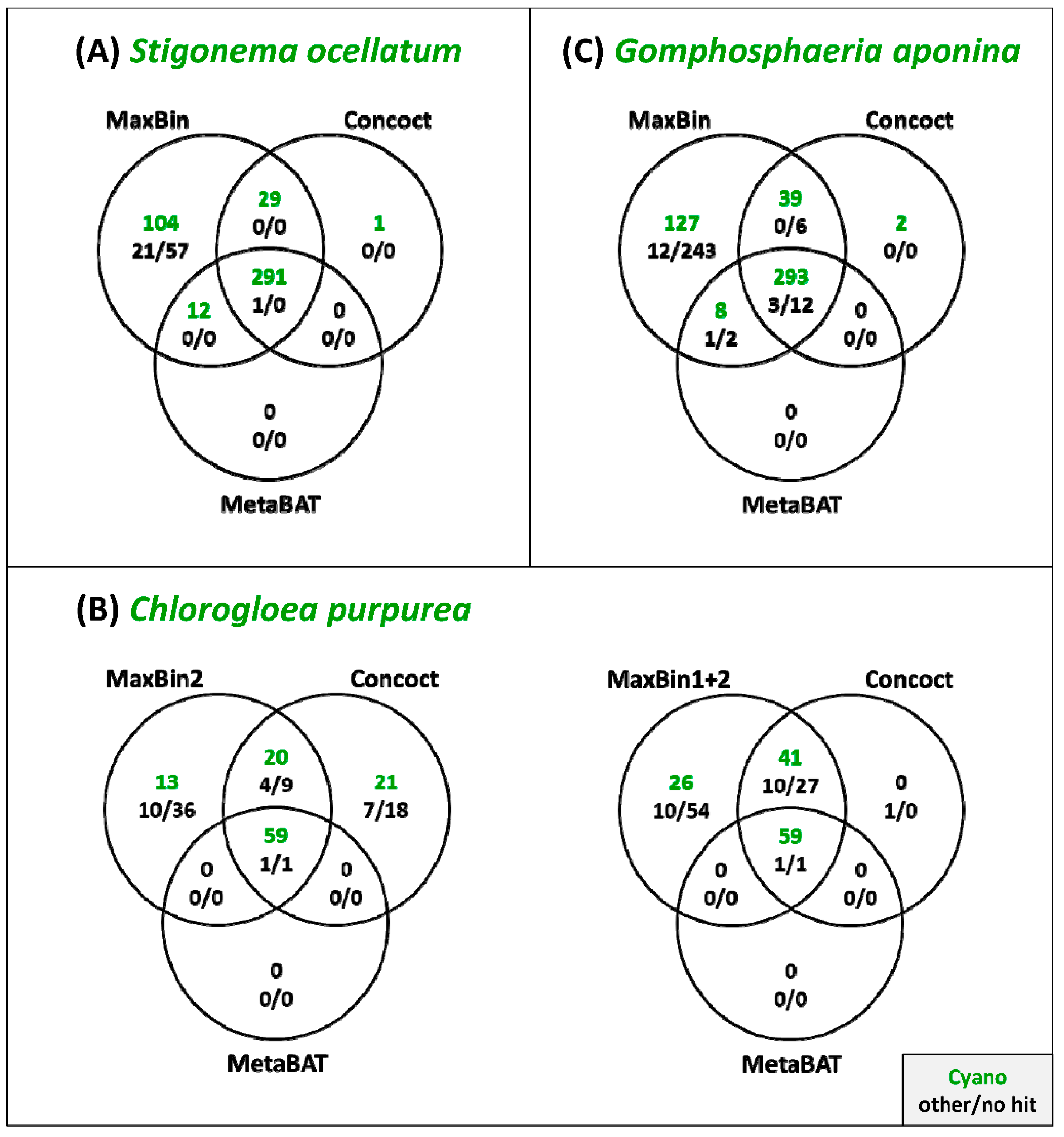
3.3. Comparison of the Binning Programs MaxBin, Concoct and MetaBAT
3.4. Genome Properties of S. ocellatum, C. purpurea and G. aponina
3.5. Discovery of Novel Bacterial Taxa in the Cyanosphere Based on the 16S-rRNA Gene
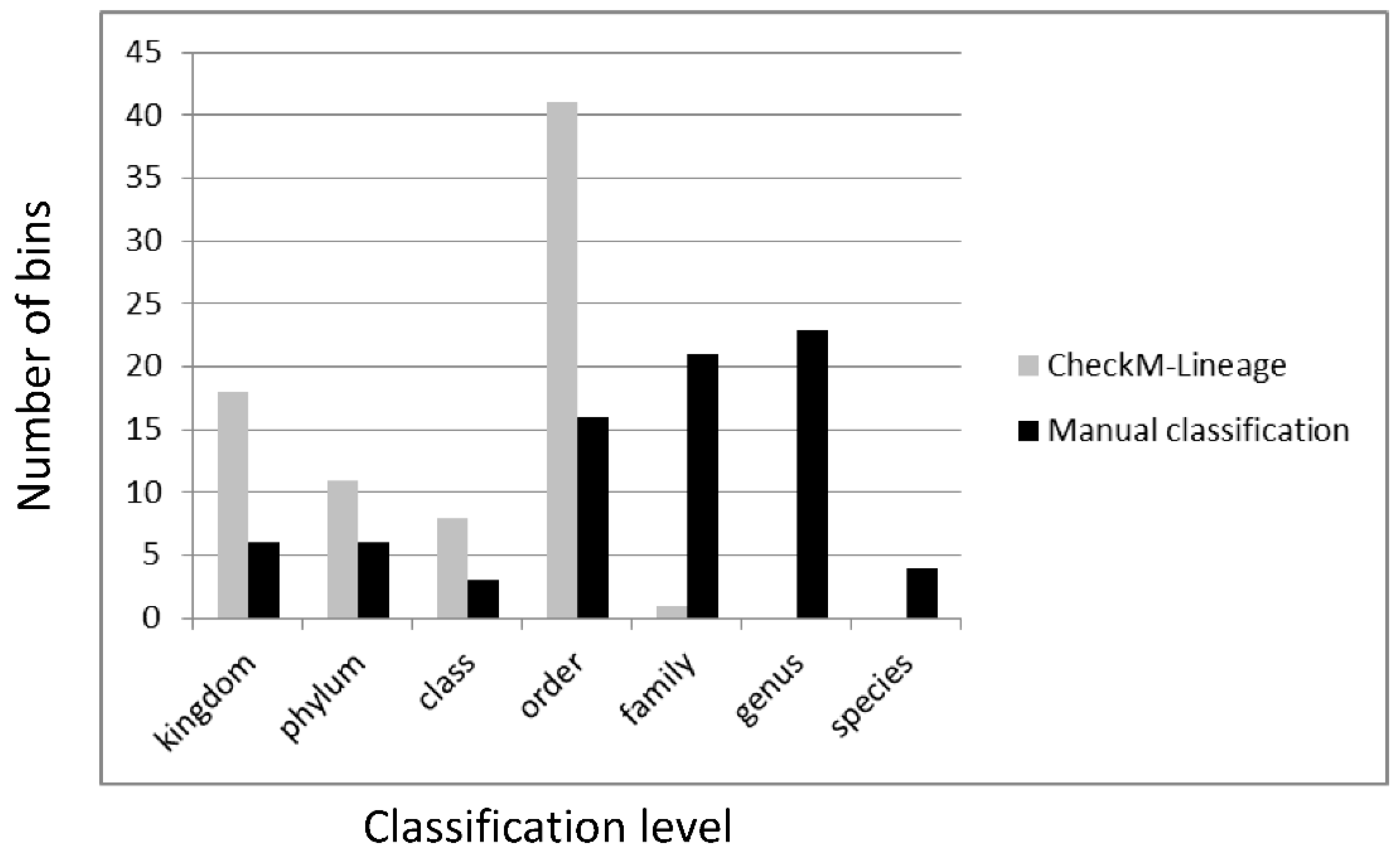
3.6. Classification of Metagenomic Bins from Non-Axenic Cyanobacteria
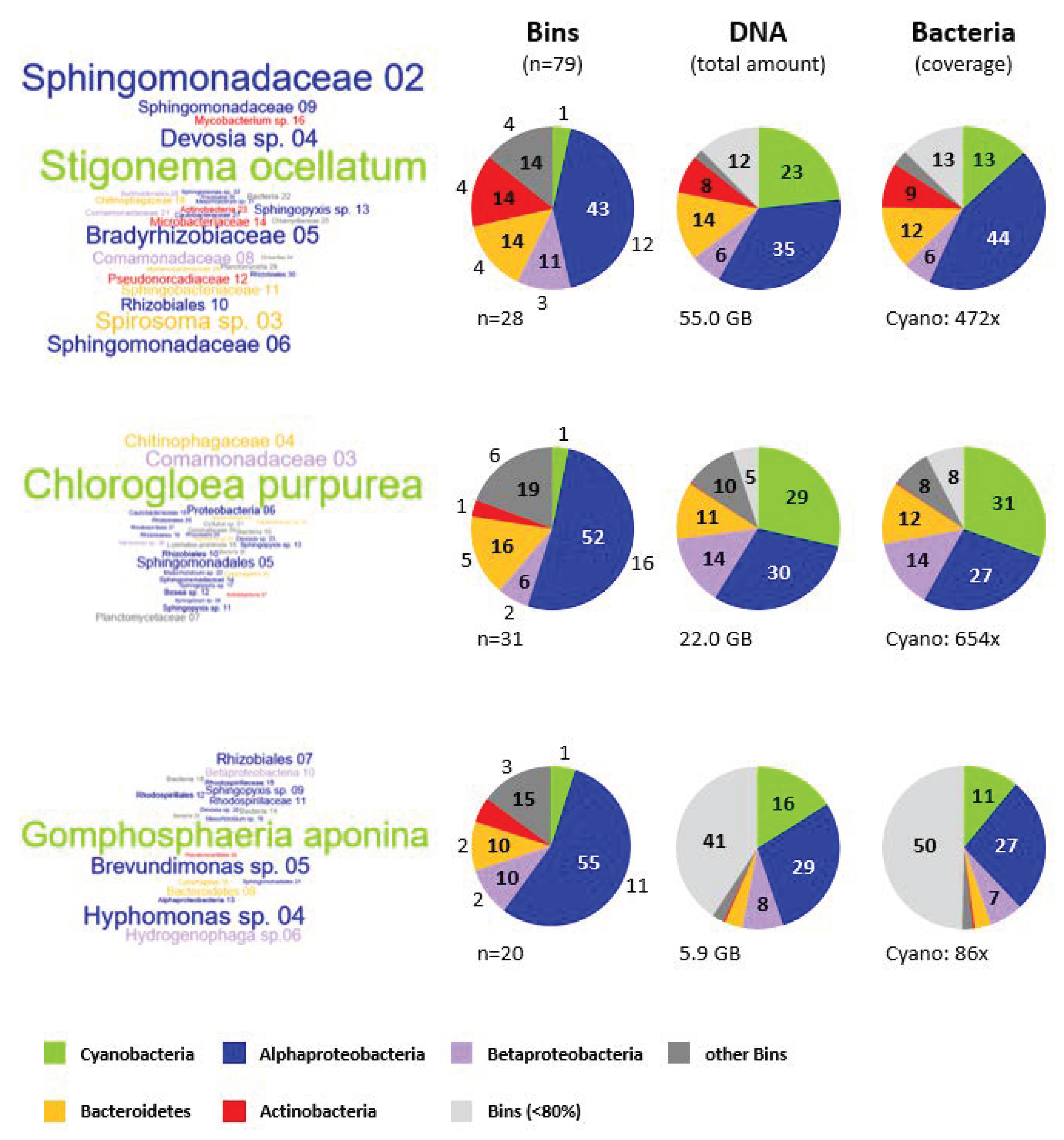
3.7. Microbial Composition of the Cyanosphere from S. ocellatum DSM 106950, C. purpurea SAG 13.99 and G. aponina DSM 107014
3.7.1. Taxonomic Affiliation of Metagenomic Bins
3.7.2. Comparison of the Cyanobacterial Metagenomes
3.8. Eukaryotic Contaminations in the Cyanosphere
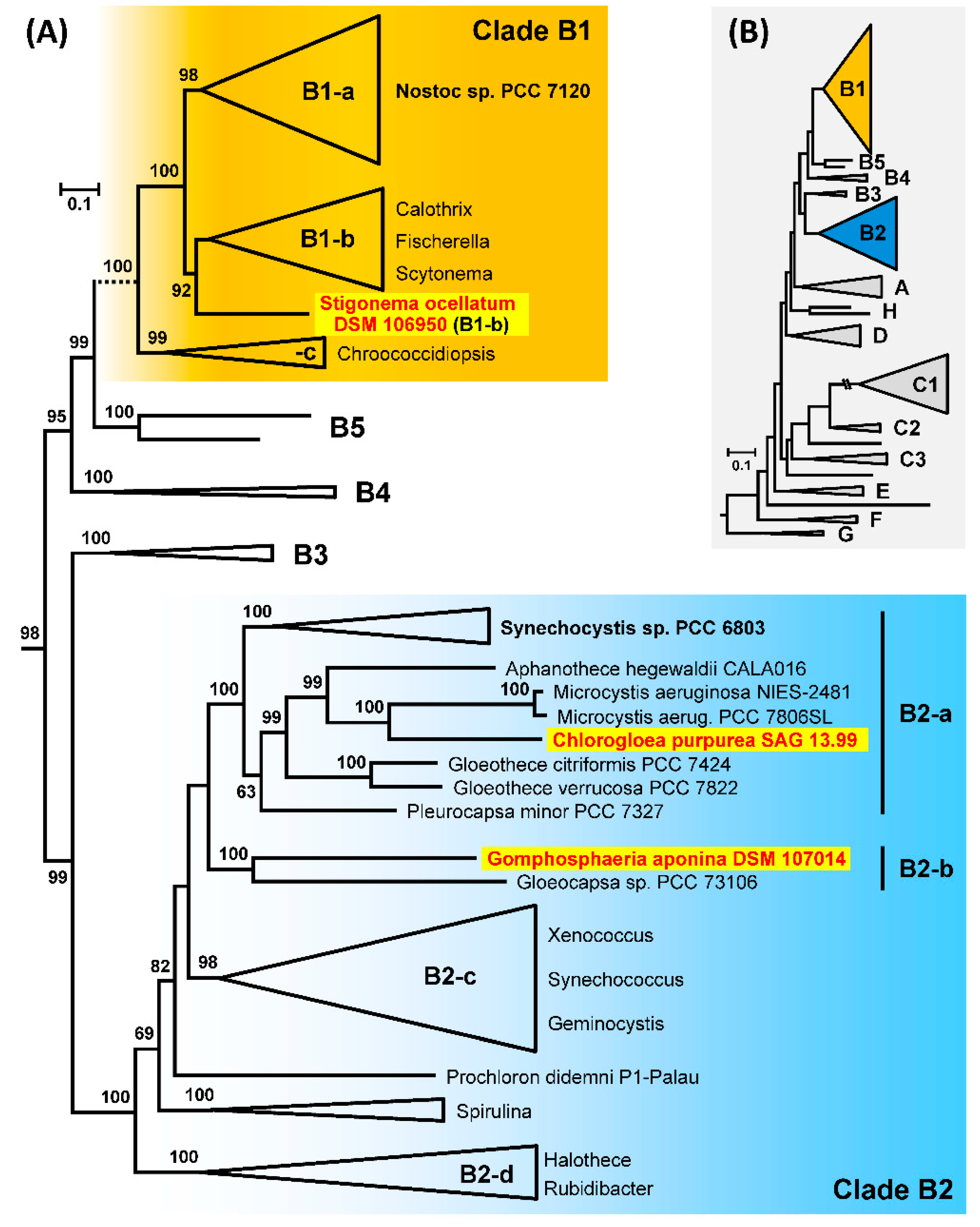
3.9. Phylogenetic Analyses of 213 Cyanobacterial Genomes
4. Discussion
4.1. Naming and Classification of Cyanobacteria
4.2. Previous Classification of the Genera Stigonema, Gomphosphaeria and Chlorogloea
4.3. Genome-Derived Phylogenies Show Incongruencies in Cyanobacterial Taxonomy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schopf, J.W. The paleobiological record of photosynthesis. Photosynth. Res. 2011, 107, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.B.; Waller, R.F.; McFadden, G.I. Plastid evolution. Annu. Rev. Plant Biol. 2008, 59, 491–517. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Ludewig, A.K.; Michael, V.; Bunk, B.; Jarek, M.; Baurain, D.; Brinkmann, H. Chromera velia, endosymbioses and the Rhodoplex hypothesis—Plastid evolution in cryptophytes, alveolates, stramenopiles, and haptophytes (CASH lineages). Genome Biol. Evol. 2014, 6, 666–684. [Google Scholar] [CrossRef] [PubMed]
- Knoll, A.H.; Nowak, M.A. The timetable of evolution. Sci. Adv. 2017, 3, e1603076. [Google Scholar] [CrossRef] [PubMed]
- Huisman, J.; Codd, G.A.; Paerl, H.W.; Ibelings, B.W.; Verspagen, J.M.H.; Visser, P.M. Cyanobacterial blooms. Nat. Rev. Microbiol. 2018, 16, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Rippka, R.; Deruelles, J.; Waterbury, J.B.; Herdman, M.; Stanier, R.Y. Generic assignments, strain histories and properties of pure cultures of Cyanobacteria. Microbiology 1979, 111, 1–61. [Google Scholar] [CrossRef]
- Christie-Oleza, J.A.; Sousoni, D.; Lloyd, M.; Armengaud, J.; Scanlan, D.J. Nutrient recycling facilitates long-term stability of marine microbial phototroph–heterotroph interactions. Nat. Microbiol. 2017, 2, 17100. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.R.; Amin, S.A.; Raina, J.B.; Stocker, R. Zooming in on the phycosphere: The ecological interface for phytoplankton-bacteria relationships. Nat. Microbiol. 2017, 2, 17065. [Google Scholar] [CrossRef] [PubMed]
- Couradeau, E.; Giraldo-Silva, A.; De Martini, F.; Garcia-Pichel, F. Spatial segregation of the biological soil crust microbiome around its foundational cyanobacterium, Microcoleus vaginatus, and the formation of a nitrogen-fixing cyanosphere. Microbiome 2019, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Oren, A.; Ventura, S. The current status of cyanobacterial nomenclature under the “prokaryotic” and the “botanical” code. Antonie Leeuwenhoek Int. J. Gen. Mol. Microbiol. 2017, 110, 1257–1269. [Google Scholar] [CrossRef]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 16048. [Google Scholar] [CrossRef] [PubMed]
- Spang, A.; Saw, J.H.; Jørgensen, S.L.; Zaremba-Niedzwiedzka, K.; Martijn, J.; Lind, A.E.; Van Eijk, R.; Schleper, C.; Guy, L.; Ettema, T.J.G. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 2015, 521, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol. 1998, 5, R245–R249. [Google Scholar] [CrossRef]
- DeLong, E.F.; Pace, N.R. Environmental diversity of bacteria and archaea. Syst. Biol. 2001, 50, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Hugenholtz, P. Exploring prokaryotic diversity in the genomic era. Genome Biol. 2002, 3, 1–8. [Google Scholar] [CrossRef][Green Version]
- Luo, C.; Tsementzi, D.; Kyrpides, N.; Read, T.; Konstantinidis, K.T. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS ONE 2012, 7, e30087. [Google Scholar] [CrossRef]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef]
- Tyson, G.W.; Chapman, J.; Hugenholtz, P.; Allen, E.E.; Ram, R.J.; Richardson, P.M.; Solovyev, V.V.; Rubin, E.M.; Rokhsar, D.S.; Banfield, J.F. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 2004, 428, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Tringe, S.G.; von Mering, C.; Kobayashi, A.; Salamov, A.A.; Chen, K.; Chang, H.W.; Podar, M.; Short, J.M.; Mathur, E.J.; Detter, J.C.; et al. Comparative metagenomics of microbial communities. Science 2005, 308, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Roller, B.R.K.; Stoddard, S.F.; Schmidt, T.M. Exploiting rRNA operon copy number to investigate bacterial reproductive strategies. Nat. Microbiol. 2016, 1, 16160. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef]
- Alneberg, J.; Bjarnason, B.S.; De Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [PubMed]
- Kunin, V.; Copeland, A.; Lapidus, A.; Mavromatis, K.; Hugenholtz, P. A bioinformatician’s guide to metagenomics. Microbiol. Mol. Biol. Rev. 2008, 72, 557–578. [Google Scholar] [CrossRef] [PubMed]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, I.C.; Dijkshoorn, L.; Whitman, W.B. Minutes of the International Committee on Systematics of Prokaryotes online discussion on the proposed use of gene sequences as type for naming of prokaryotes, and outcome of vote. Int. J. Syst. Evol. Microbiol. 2020, 70, 4416–4417. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.E.; Freudenstein, J.; Gribaldo, S.; Hatzenpichler, R.; Hugenholtz, P.; Kämpfer, P.; Konstantinidis, K.T.; Lane, C.E.; Papke, R.T.; Parks, D.H.; et al. Roadmap for naming uncultivated Archaea and Bacteria. Nat. Microbiol. 2020, 5, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Alvarenga, D.O.; Fiore, M.F.; Varani, A.M. A metagenomic approach to cyanobacterial genomics. Front. Microbiol. 2017, 8, 809. [Google Scholar] [CrossRef] [PubMed]
- Grim, S.L.; Dick, G.J. Photosynthetic versatility in the genome of Geitlerinema sp. PCC 9228 (Formerly Oscillatoria limnetica ’solar lake’), a model anoxygenic photosynthetic cyanobacterium. Front. Microbiol. 2016, 7, 1546. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, C.B.; Otten, T.G.; Brown, N.M.; Dreher, T.W. Towards long-read metagenomics: Complete assembly of three novel genomes from bacteria dependent on a diazotrophic cyanobacterium in a freshwater lake co-culture. Stand. Genom. Sci. 2017, 12, 1–16. [Google Scholar] [CrossRef]
- Cornet, L.; Bertrand, A.R.; Hanikenne, M.; Javaux, E.J.; Wilmotte, A.; Baurain, D. Metagenomic assembly of new (sub)arctic Cyanobacteria and their associated microbiome from non-axenic cultures. Microb. Genom. 2018, 1–15. [Google Scholar] [CrossRef]
- Will, S.E.; Henke, P.; Boedeker, C.; Huang, S.; Brinkmann, H.; Rohde, M.; Jarek, M.; Friedl, T.; Seufert, S.; Schumacher, M.; et al. Day and night: Metabolic profiles and evolutionary relationships of six axenic non-marine cyanobacteria. Genome Biol. Evol. 2019, 11, 270–294. [Google Scholar] [CrossRef] [PubMed]
- Cornet, L.; Meunier, L.; Van Vlierberghe, M.; Leonard, R.R.; Durieu, B.; Lara, Y.; Misztak, A.; Sirjacobs, D.; Javaux, E.J.; Philippe, H.; et al. Consensus assessment of the contamination level of publicly available cyanobacterial genomes. PLoS ONE 2018, 13, e0200323. [Google Scholar] [CrossRef] [PubMed]
- Komárek, J.; Kaštovský, J.; Mareš, J.; Johansen, J.R. Taxonomic classification of cyanoprokaryotes (cyanobacteria genera) 2014, using a polyphasic approach. Preslia 2014, 86, 295–335. [Google Scholar]
- Cornet, L.; Wilmotte, A.; Javaux, E.J.; Baurain, D. A constrained SSU-rRNA phylogeny reveals the unsequenced diversity of photosynthetic Cyanobacteria (Oxyphotobacteria). BMC Res. Notes 2018, 11, 435. [Google Scholar] [CrossRef] [PubMed]
- Aronesty, E. Command-line Tools for Processing Biological Sequencing Data. Available online: https://github.com/ExpressionAnalysis/ea-utils (accessed on 9 December 2019).
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Tanizawa, Y.; Fujisawa, T.; Nakamura, Y. DFAST: A flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 2018, 34, 1037–1039. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Philippe, H. MUST, a computer package of Management Utilities for Sequences and Trees. Nucleic Acids Res. 1993, 21, 5264–5272. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Shi, L.; Huang, Y.; Zhang, M.; Yu, Y.; Lu, Y.; Kong, F. Bacterial community dynamics and functional variation during the long-term decomposition of cyanobacterial blooms in-vitro. Sci. Total Environ. 2017, 598, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Eraqi, W.A.; Elrakaiby, M.T.; Megahed, S.A.; Yousef, N.H.; Elshahed, M.S.; Yassin, A.S. The Nile River microbiome reveals a remarkably stable community between wet and dry seasons, and sampling sites, in a large urban metropolis (Cairo, Egypt). Omics J. Integr. Biol. 2018, 22, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Parsons, R.J.; Breitbart, M.; Lomas, M.W.; Carlson, C.A. Ocean time-series reveals recurring seasonal patterns of virioplankton dynamics in the northwestern Sargasso Sea. ISME J. 2012, 6, 273–284. [Google Scholar] [CrossRef]
- Blandenier, Q.; Seppey, C.V.W.; Singer, D.; Vlimant, M.; Simon, A.; Duckert, C.; Lara, E. Mycamoeba gemmipara nov. gen., nov. sp., the first cultured member of the environmental Dermamoebidae clade LKM74 and its unusual life cycle. J. Eukaryot. Microbiol. 2017, 64, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Dyková, I.; Boháčová, L.; Fiala, I.; Macháčková, B.; Pecková, H.; Dvořáková, H. Amoebae of the genera Vannella Bovee, 1965 and Platyamoeba Page, 1969 isolated from fish and their phylogeny inferred from SSU rRNA gene and ITS sequences. Eur. J. Protistol. 2005, 41, 219–230. [Google Scholar] [CrossRef]
- Sogin, M.L.; Silberman, J.D. Evolution of the protists and protistan parasites from the perspective of molecular systematics. Int. J. Parasitol. 1998, 28, 11–20. [Google Scholar] [CrossRef]
- De Jonckheere, J.; Murase, J.; Opperdoes, F.R. A new thermophilic heterolobosean amoeba, Fumarolamoeba ceborucoi, gen. nov., sp. nov., isolated near a fumarole at a volcano in Mexico. Acta Protozool. 2011, 50, 43–50. [Google Scholar]
- Shih, P.M.; Wu, D.; Latifi, A.; Axen, S.D.; Fewer, D.P.; Talla, E.; Calteau, A.; Cai, F.; Tandeau de Marsac, N.; Rippka, R.; et al. Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 1053–1058. [Google Scholar] [CrossRef]
- Harke, M.J.; Steffen, M.M.; Gobler, C.J.; Otten, T.G.; Wilhelm, S.W.; Wood, S.A.; Paerl, H.W. A review of the global ecology, genomics, and biogeography of the toxic cyanobacterium, Microcystis spp. Harmful Algae 2016, 54, 4–20. [Google Scholar] [CrossRef]
- Gugger, M.F.; Hoffmann, L. Polyphyly of true branching cyanobacteria (Stigonematales). Int. J. Syst. Evol. Microbiol. 2004, 54, 349–357. [Google Scholar] [CrossRef]
- Bauersachs, T.; Miller, S.R.; Gugger, M.; Mudimu, O.; Friedl, T.; Schwark, L. Heterocyte glycolipids indicate polyphyly of stigonematalean cyanobacteria. Phytochemistry 2019, 166. [Google Scholar] [CrossRef] [PubMed]
- Oren, A.; Tindall, B.J. Nomenclature of the cyanophyta/cyanobacteria/cyanoprokaryotes under the International Code of Nomenclature of Prokaryotes. Arch. Hydrobiol. Suppl. Algol. Stud. 2005, 117, 39–52. [Google Scholar] [CrossRef]
- Hördt, A.; López, M.G.; Meier-Kolthoff, J.P.; Schleuning, M.; Weinhold, L.M.; Tindall, B.J.; Gronow, S.; Kyrpides, N.C.; Woyke, T.; Göker, M. Analysis of 1,000+ type-strain genomes substantially improves taxonomic classification of Alphaproteobacteria. Front. Microbiol. 2020, 11, 468. [Google Scholar] [CrossRef]
- García-López, M.; Meier-Kolthoff, J.P.; Tindall, B.J.; Gronow, S.; Woyke, T.; Kyrpides, N.C.; Hahnke, R.L.; Göker, M. Analysis of 1,000 type-strain genomes improves taxonomic classification of Bacteroidetes. Front. Microbiol. 2019, 10, 2083. [Google Scholar] [CrossRef] [PubMed]
- Mooney, K.M.; Hamilton, J.T.G.; Floyd, S.D.; Robert, H.F.; Elliott, C.T. Initial studies on the occurence of cyanobacteria and microcystins in Irish lakes. Environ. Toxicol. 2011, 26, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.S.P.; Genuário, D.B.; Vaz, M.G.M.V.; Fiore, M.F. Phylogeny of culturable cyanobacteria from Brazilian mangroves. Syst. Appl. Microbiol. 2014, 37, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, F.; Tschoeke, D.A.; Thompson, F.; Thompson, C. Comparative genomics of Synechococcus and proposal of the new genus Parasynechococcus. PeerJ 2016, 2016, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Salazar, V.W.; Tschoeke, D.A.; Swings, J.; Cosenza, C.A.; Mattoso, M.; Thompson, C.C.; Thompson, F.L. A new genomic taxonomy system for the Synechococcus collective. Environ. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stigonema ocellatum | Chlorogloea purpurea | Gomphosphaeria aponina | |
|---|---|---|---|
| Morphology | |||
| Cells | filamentous | unicellular | unicellular-colonial |
| Cell shape | uniseriate | spherical | heart-shaped cells |
| Sheath | + | + | + |
| Branching | T-branching | - | - |
| Heterocysts | intercalary | - | - |
| Phylogeny | |||
| Clade 1 | B1-b | B2-a | B2-b |
| Order | Nostocales | Chroococcales | Chroococcales |
| Family | Stigonemataceae | Entophysalidaceae | Gomphosphaeriaceae |
| Genome | |||
| MAG status | high-quality draft | high-quality draft 2 | high-quality draft 2 |
| Contigs | 509 | 228 | 749 |
| Size (Mb) | 10.35 | 4.74 | 5.34 |
| G + C | 43.8 | 45.3 | 38.9 |
| Accession number | JADQBA000000000 | JADQBB000000000 | JADQBC000000000 |
| Isolation | |||
| Origin | Allgäu, Germany | Serra da Estrela, Portugal | Neusiedlersee, Austria |
| Habitat | freshwater | freshwater | freshwater |
| Locality | Sphagnum bog | flowing water | benthic on Phragmites |
| Scientist | D. Mollenhauer | M. F. Santos | E. Kusel-Fetzmann |
| Year | 1970 | 1981 | 1985 |
| Publications | Gugger and Hoffmann (2004) Bauersachs et al. (2019) | - | - |
| Collection ID | |||
| DSM | 106950 | - | 107014 |
| SAG | 48.90 | 13.99 | 52.96 |
| Attribute | Stigonema ocellatum DSM 106950 | Chlorogloea purpurea SAG 13.99 | Gomphosphaeria aponina DSM 107014 | |||
|---|---|---|---|---|---|---|
| Value | % of Total | Value | % of Total | Value | % of Total | |
| Genome size (bp) | 10,354,468 | 100.00 | 4,737,903 | 100.00 | 5,337,155 | 100.00 |
| DNA coding (bp) | 7,983,295 | 77.10 | 4,140,927 | 87.40 | 4,360,456 | 81.70 |
| DNA G + C (bp) | 4,535,257 | 43.80 | 2,146,270 | 45.30 | 2,081,490 | 39.00 |
| Contigs | 509 | 228 | 749 | |||
| Total genes | 8824 | 100.00 | 4429 | 100.00 | 5305 | 100.00 |
| Protein coding genes | 8701 | 98.61 | 4384 | 98.98 | 5235 | 98.68 |
| RNA genes | 3 | 0.03 | 2 | 0.05 | 2 | 0.04 |
| Pseudo genes | 120 | 1.36 | 43 | 0.97 | 68 | 1.28 |
| Genes with function prediction | 2826 | 32.03 | 1931 | 43.60 | 1846 | 34.80 |
| Genes assigned to COGs | 4011 | 45.46 | 2250 | 50.80 | 2413 | 45.49 |
| Genes with Pfam domains | 5985 | 67.83 | 3249 | 73.36 | 3751 | 70.71 |
| Genes with signal peptides | 664 | 7.52 | 350 | 7.90 | 446 | 8.41 |
| Genes with transmembrane helices (≥3) | 708 | 8.02 | 449 | 10.14 | 378 | 7.13 |
| CRISPR repeats | 13 | 0.15 | 10 | 0.23 | 19 | 0.36 |
| 16S-rRNA Gene (BLASTN) | |
| Pro | Contra |
| (1) Gold standard for prokaryotic taxonomy | (1) Frequent lack of marker gene in the bins * |
| (2) Well-curated reference sequences from all type strains | (2) Wrong marker gene(s) in the bins * |
| (3) Large set of reference sequences (isolates, environment) | (3) Comparably poor taxonomic resolution |
| (4) Thresholds for delineation of species and higher order taxa | |
| RpoB Protein (BLASTP) | |
| Pro | Contra |
| (1) Representative codon usage and nucleotide composition (essential protein) | (1) No general thresholds for species delineation |
| (2) Coverage diagnostic for the genome (single-copy gene) | (2) Inconclusive classification of bins with a high contamination level |
| (3) Large protein with good taxonomic resolution | |
| (4) Present in most bins with a completeness > 90% | |
| Text Mining (BLASTN) | |
| Pro | Contra |
| (1) Rapid assessment of metagenomes | (1) Taxonomic resolution limited to genus level |
| (2) Applicable for incomplete and contaminated bins | |
| (3) Reliable identification of the dominant (primary) genome | |
| (4) Detection of plasmid-related bins lacking any marker gene | |
| (5) Identification of eukaryotic bins | |
| (A) Stigonema ocellatum DSM 106950 | ||||||||||
| Binning Results | Classification 1 | |||||||||
| bin | Compl. | Contam. | Contigs | Genome (bp) | Coverage | 16S | RpoB | Text | RpoB & Text Mining | Tax. |
| 01 | 99.28 | 2.35 | 515 | 10,519,008 | 471.84 | yes/no | “yes” | “yes” | S. ocellatum 2 | s |
| 02 | 96.00 | 3.33 | 106 | 3,017,240 | 415.54 | no | yes | yes | Sphingomonadaceae | f |
| 03 | 100.0 | 0.74 | 184 | 7,762,495 | 244.90 | false | yes | yes | Spirosoma sp. | g |
| 04 | 80.80 | 5.34 | 36 | 4,445,593 | 244.15 | x | yes | yes | Devosia sp. | g |
| 05 | 86.21 | 33.89 | 69 | 4,652,777 | 242.25 | no | yes | yes | Bradyrhizobiaceae | f |
| 06 | 99.59 | 8.90 | 28 | 4,785,235 | 219.72 | x | yes | yes | Sphingomonadaceae | f |
| 08 | 98.44 | 2.23 | 36 | 6,359,601 | 149.04 | x | x | yes | Comamonadaceae | f |
| 09 | 93.10 | 36.99 | 134 | 7,299,278 | 143.94 | x | yes/no | mult. | Sphingomonadaceae | f |
| 10 | 83.92 | 3.82 | 94 | 8,160,230 | 142.77 | x | yes | yes | Rhizobiales | o |
| 11 | 98.10 | 0.71 | 62 | 4,257,773 | 122.35 | false | yes | yes/no | Sphingobacteriaceae | f |
| 12 | 99.17 | 2.73 | 79 | 7,053,193 | 106.66 | x | yes | yes | Pseudonorcadiaceae | f |
| 13 | 99.66 | 0.63 | 53 | 3,680,574 | 98.53 | x | yes | yes | Sphingopyxis sp. | g |
| 14 | 97.81 | 0.27 | 28 | 2,624,238 | 94.72 | yes | yes | yes | Microbacteriaceae | f |
| 16 | 100.00 | 0.13 | 33 | 6,260,391 | 74.38 | x | yes | yes | Mycobacterium sp. | g |
| 18 | 99.51 | 1.08 | 49 | 6,025,211 | 61.24 | yes | yes | yes/no | Chitinophagaceae | f |
| 21 | 92.52 | 2.80 | 114 | 7,675,694 | 41.20 | x | x | yes | Comamonadaceae | f |
| 22 | 98.46 | 2.72 | 19 | 4,521,530 | 41.19 | x | x | yes/no | Bacteria | k |
| 23 | 95.26 | 0.00 | 148 | 3,446,731 | 41.02 | yes | yes | yes | Actinobacteria | p |
| (B) Chlorogloea purpurea SAG 13.99 | ||||||||||
| Binning Results | Classification 1 | |||||||||
| bin | Compl. | Contam. | Contigs | Genome (bp) | Coverage | 16S | RpoB | Text | RpoB & Text Mining | Tax. |
| 01 + 02 * | 100.00 | 0.29 | 186 | 4,595,485 | 654.42 | x | “yes” | “Yes” | C. purpurea2 | s |
| 03 | 85.86 | 5.70 | 131 | 4,964,628 | 288.68 | x | x | yes | Comamonadaceae | f |
| 04 | 97.04 | 0.52 | 37 | 4,500,700 | 230.28 | yes | yes | yes | Chitinophagaceae | f |
| 05 | 81.59 | 14.77 | 93 | 4,443,934 | 150.84 | false | x | yes/no | Sphingomonadales | o |
| 06 | 98.12 | 30.88 | 130 | 6,184,863 | 105.77 | x | mult. | mult. | Proteobacteria | p |
| 07 | 91.03 | 2.30 | 81 | 7,916,241 | 87.81 | x | yes | yes/yes | Planctomycetaceae | f |
| 10 | 89.39 | 25.52 | 79 | 8,815,014 | 60.85 | x | x | yes/no | Rhizobiales | o |
| 11 | 85.54 | 10.13 | 48 | 4,001,378 | 52.94 | x | yes | yes | Sphingopyxis sp. | g |
| 12 | 93.73 | 34.01 | 91 | 8,062,010 | 45.99 | x | yes | yes | Bosea sp. | g |
| 13 | 89.40 | 17.80 | 58 | 3,664,549 | 37.48 | x | false | yes | Sphingopyxis sp. | g |
| 14 | 99.09 | 1.75 | 36 | 3,405,585 | 31.73 | yes | yes | yes | Sphingomonadaceae | f |
| 15 | 97.62 | 0.98 | 33 | 4,008,507 | 28.58 | no | yes | yes/no | Bacteria | k |
| 16 | 100.0 | 13.79 | 72 | 5,989,586 | 25.63 | yes | yes | yes/no | Luteitalea pratensis | s |
| (C) Gomphosphaeria aponina DSM 107014 | ||||||||||
| Binning Results | Classification 1 | |||||||||
| bin | Compl. | Contam. | Contigs | Genome (bp) | Coverage | 16S | RpoB | Text | RpoB & Text Mining | Tax. |
| 03 | 97.82 | 0.11 | 746 | 5,354,489 | 85.91 | x | “yes” | “Yes” | G. aponina 2 | s |
| 04 | 100.00 | 5.55 | 277 | 4,200,065 | 58.22 | yes | yes | mult. | Hyphomonas sp. | g |
| 05 | 94.48 | 4.22 | 247 | 2,796,749 | 49.08 | yes/no | yes | yes | Brevundimonas sp. | g |
| 06 | 99.53 | 1.40 | 378 | 4,670,292 | 34.15 | x | yes | yes | Hydrogenophaga sp. | g |
| 07 | 98.54 | 2.51 | 174 | 3,619,276 | 29.06 | false | yes | yes | Rhizobiales | o |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marter, P.; Huang, S.; Brinkmann, H.; Pradella, S.; Jarek, M.; Rohde, M.; Bunk, B.; Petersen, J. Filling the Gaps in the Cyanobacterial Tree of Life—Metagenome Analysis of Stigonema ocellatum DSM 106950, Chlorogloea purpurea SAG 13.99 and Gomphosphaeria aponina DSM 107014. Genes 2021, 12, 389. https://doi.org/10.3390/genes12030389
Marter P, Huang S, Brinkmann H, Pradella S, Jarek M, Rohde M, Bunk B, Petersen J. Filling the Gaps in the Cyanobacterial Tree of Life—Metagenome Analysis of Stigonema ocellatum DSM 106950, Chlorogloea purpurea SAG 13.99 and Gomphosphaeria aponina DSM 107014. Genes. 2021; 12(3):389. https://doi.org/10.3390/genes12030389
Chicago/Turabian StyleMarter, Pia, Sixing Huang, Henner Brinkmann, Silke Pradella, Michael Jarek, Manfred Rohde, Boyke Bunk, and Jörn Petersen. 2021. "Filling the Gaps in the Cyanobacterial Tree of Life—Metagenome Analysis of Stigonema ocellatum DSM 106950, Chlorogloea purpurea SAG 13.99 and Gomphosphaeria aponina DSM 107014" Genes 12, no. 3: 389. https://doi.org/10.3390/genes12030389
APA StyleMarter, P., Huang, S., Brinkmann, H., Pradella, S., Jarek, M., Rohde, M., Bunk, B., & Petersen, J. (2021). Filling the Gaps in the Cyanobacterial Tree of Life—Metagenome Analysis of Stigonema ocellatum DSM 106950, Chlorogloea purpurea SAG 13.99 and Gomphosphaeria aponina DSM 107014. Genes, 12(3), 389. https://doi.org/10.3390/genes12030389