
Embryonic Kidney Development, Stem Cells and the Origin of Wilms Tumor


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
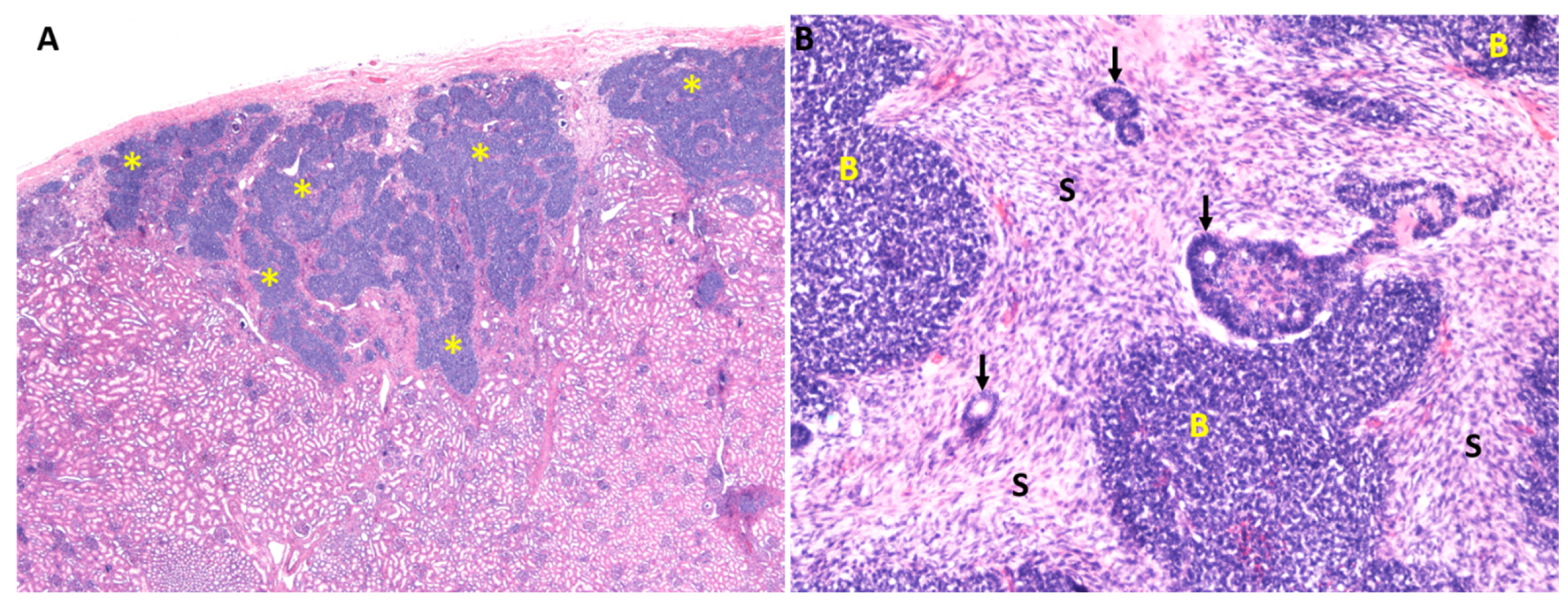
2. Wilms Tumor
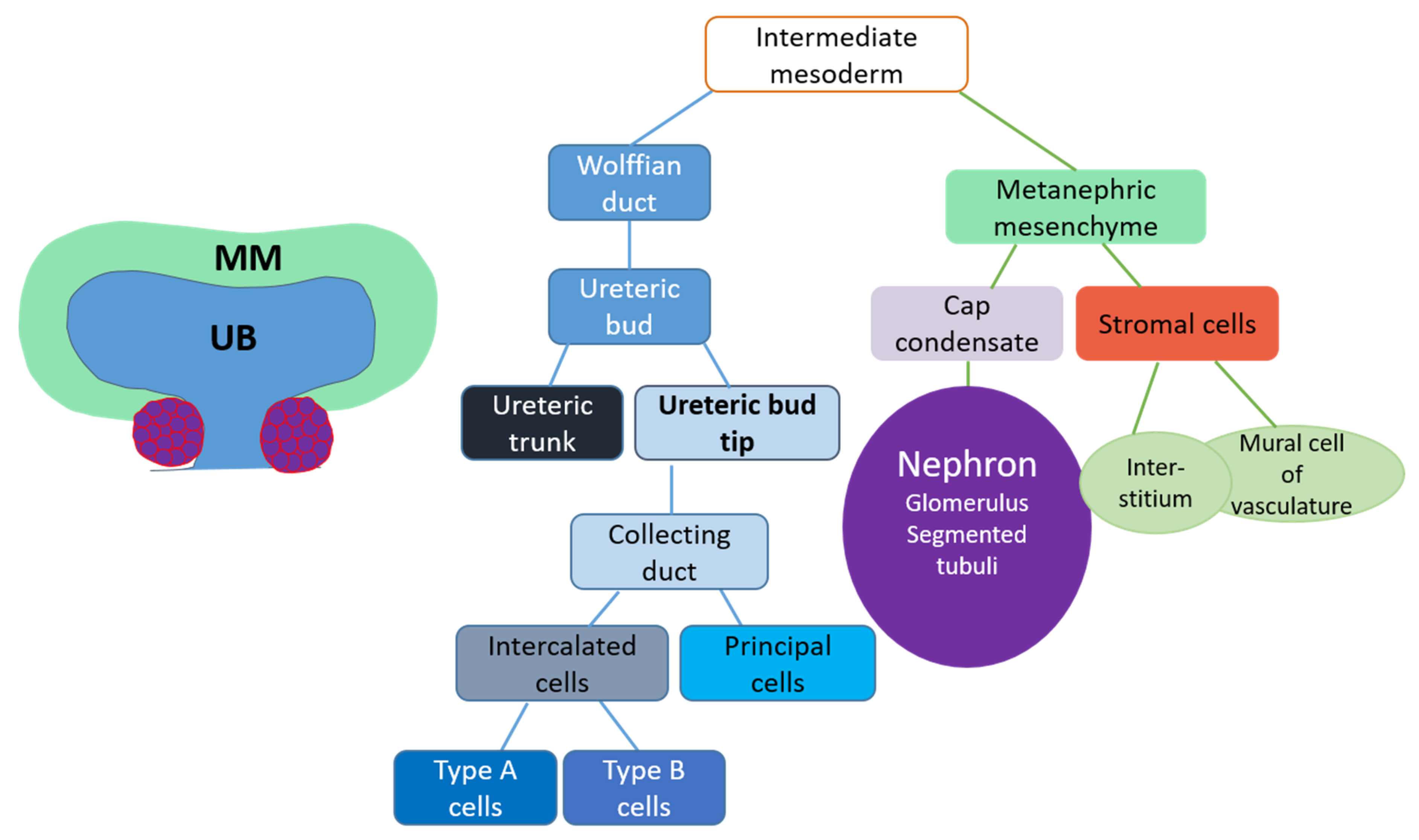
3. Embryonic Kidney
3.1. Kidney Induction
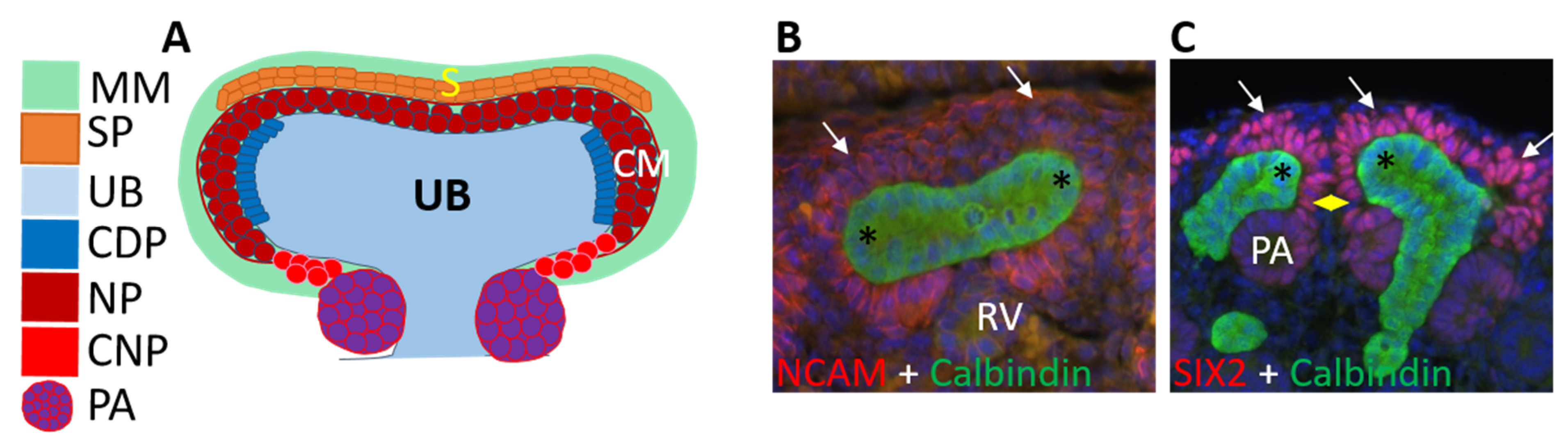
3.2. Ureteric Bud Branching Morphogenesis Orchestrates Embryonic Kidney Growth and Nephron Formation
4. Renal Progenitor Populations
5. Collecting Duct Progenitors
GDNF/RET Signaling Regulates Collecting Duct Progenitors through MAPK/ERK Activity
6. Stromal Progenitor Cells
7. Nephron Progenitors
7.1. Molecular Determinants of Nephron Progenitors
7.2. Nephron Progenitor Maintenance Depends on Classical Signaling Pathway Activities
7.3. The Wnt Pathway
7.4. FGF induced Receptor Tyrosine Kinase Signaling
7.5. Permanent Loss of Nephron Progenitors Terminates Renal Development
8. The Cause of Wilms Tumors
9. The Origins of Wilms Tumors
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pleniceanu, O.; Omer, D.; Harari-Steinberg, O.; Dekel, B. Renal lineage cells as a source for renal regeneration. Pediatr. Res. 2018, 83, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.; Nishinakamura, R. How to rebuild the kidney: Recent advances in kidney organoids. J. Biochem. 2019, 166, 7–12. [Google Scholar] [CrossRef]
- McMahon, A.P. Development of the Mammalian Kidney. Curr. Top. Dev. Biol. 2016, 117, 31–64. [Google Scholar] [CrossRef]
- Kurtzeborn, K.; Kwon, H.N.; Kuure, S. MAPK/ERK Signaling in Regulation of Renal Differentiation. Int. J. Mol. Sci. 2019, 20, 1779. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Valerius, M.T.; Mugford, J.W.; Carroll, T.J.; Self, M.; Oliver, G.; McMahon, A.P. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 2008, 3, 169–181. [Google Scholar] [CrossRef]
- Lin, E.E.; Sequeira-Lopez, M.L.S.; Gomez, R.A. RBP-J in FOXD1+renal stromal progenitors is crucial for the proper development and assembly of the kidney vasculature and glomerular mesangial cells. Am. J. Physiol. Renal. 2014, 306, F249–F258. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, P.; Pritchard-Jones, K.; Charlton, J. The yin and yang of kidney development and Wilms’ tumors. Genes Dev. 2015, 29, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Leslie, S.W.; Sajjad, H.; Murphy, P.B. Wilms Tumor (Nephroblastoma). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Leslie, S.W.; Murphy, P.B. Cancer, Wilms (Nephroblastoma). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Knudson, A.G., Jr.; Strong, L.C. Mutation and cancer: A model for Wilms’ tumor of the kidney. J. Natl. Cancer Inst. 1972, 48, 313–324. [Google Scholar]
- Saxen, L.; Sariola, H. Early organogenesis of the kidney. Pediatr. Nephrol. 1987, 1, 385–392. [Google Scholar] [CrossRef]
- Saxen, L.; Vainio, S.; Jalkanen, M.; Lehtonen, E. Intercellular adhesion and induction of epithelialization in the metanephric mesenchyme. Cell Differ. Dev. 1988, 25, 111–117. [Google Scholar] [CrossRef]
- Cebrian, C.; Asai, N.; D’Agati, V.; Costantini, F. The number of fetal nephron progenitor cells limits ureteric branching and adult nephron endowment. Cell Rep. 2014, 7, 127–137. [Google Scholar] [CrossRef]
- Kuure, S.; Sariola, H. Mouse Models of Congenital Kidney Anomalies. Adv. Exp. Med. Biol. 2020, 1236, 109–136. [Google Scholar] [CrossRef]
- Saxen, L. Organogenesis of the Kidney; Cambridge University Press: Cambridge, UK, 1987. [Google Scholar]
- Taguchi, A.; Kaku, Y.; Ohmori, T.; Sharmin, S.; Ogawa, M.; Sasaki, H.; Nishinakamura, R. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 2014, 14, 53–67. [Google Scholar] [CrossRef]
- Stewart, K.; Bouchard, M. Coordinated cell behaviours in early urogenital system morphogenesis. Semin. Cell Dev. Biol. 2014, 36, 13–20. [Google Scholar] [CrossRef]
- Oxburgh, L. Kidney Nephron Determination. Annu. Rev. Cell Dev. Biol. 2018, 34, 427–450. [Google Scholar] [CrossRef] [PubMed]
- Hatini, V.; Huh, S.O.; Herzlinger, D.; Soares, V.C.; Lai, E. Essential role of stromal mesenchyme in kidney morphogenesis revealed by targeted disruption of Winged Helix transcription factor BF-2. Genes Dev. 1996, 10, 1467–1478. [Google Scholar] [CrossRef]
- Hum, S.; Rymer, C.; Schaefer, C.; Bushnell, D.; Sims-Lucas, S. Ablation of the renal stroma defines its critical role in nephron progenitor and vasculature patterning. PLoS ONE 2014, 9, e88400. [Google Scholar] [CrossRef]
- Zhang, H.; Bagherie-Lachidan, M.; Badouel, C.; Enderle, L.; Peidis, P.; Bremner, R.; Kuure, S.; Jain, S.; McNeill, H. FAT4 Fine-Tunes Kidney Development by Regulating RET Signaling. Dev. Cell 2019, 48, 780–792.e784. [Google Scholar] [CrossRef] [PubMed]
- Batourina, E.; Gim, S.; Bello, N.; Shy, M.; Clagett-Dame, M.; Srinivas, S.; Costantini, F.; Mendelsohn, C. Vitamin A controls epithelial/mesenchymal interactions through Ret expression. Nat. Genet. 2001, 27, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.C.; Rivera-Reyes, R.; Englert, C.; Kispert, A. Expansion of the renal capsular stroma, ureteric bud branching defects and cryptorchidism in mice with Wilms tumor 1 gene deletion in the stromal compartment of the developing kidney. J. Pathol. 2020, 252, 290–303. [Google Scholar] [CrossRef]
- Mohamed, T.; Sequeira-Lopez, M.L.S. Development of the renal vasculature. Semin. Cell Dev. Biol. 2019, 91, 132–146. [Google Scholar] [CrossRef]
- Herzlinger, D.; Hurtado, R. Patterning the renal vascular bed. Semin. Cell Dev. Biol. 2014, 36, 50–56. [Google Scholar] [CrossRef]
- Sariola, H.; Holm, K.; Henke, F.S. Early innervation of the metanephric kidney. Development 1988, 104, 589–599. [Google Scholar]
- Sariola, H.; Holm, S.K.; Henke, F.S. The effect of neuronal cells on kidney differentiation. Int. J. Dev. Biol. 1989, 33, 149–155. [Google Scholar] [PubMed]
- Burnstock, G.; Loesch, A. Sympathetic innervation of the kidney in health and disease: Emphasis on the role of purinergic cotransmission. Auton. Neurosci. 2017, 204, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Munro, D.A.D.; Hohenstein, P.; Davies, J.A. Cycles of vascular plexus formation within the nephrogenic zone of the developing mouse kidney. Sci. Rep. 2017, 7, 3273. [Google Scholar] [CrossRef]
- Munro, D.A.D.; Wineberg, Y.; Tarnick, J.; Vink, C.S.; Li, Z.; Pridans, C.; Dzierzak, E.; Kalisky, T.; Hohenstein, P.; Davies, J.A. Macrophages restrict the nephrogenic field and promote endothelial connections during kidney development. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Sims-Lucas, S.; Schaefer, C.; Bushnell, D.; Ho, J.; Logar, A.; Prochownik, E.; Gittes, G.; Bates, C.M. Endothelial Progenitors Exist within the Kidney and Lung Mesenchyme. PLoS ONE 2013, 8, e65993. [Google Scholar] [CrossRef]
- Alabi, R.O.; Farber, G.; Blobel, C.P. Intriguing Roles for Endothelial ADAM10/Notch Signaling in the Development of Organ-Specific Vascular Beds. Physiol. Rev. 2018, 98, 2025–2061. [Google Scholar] [CrossRef]
- Mukherjee, E.; Maringer, K.; Papke, E.; Bushnell, D.; Schaefer, C.; Kramann, R.; Ho, J.; Humphreys, B.D.; Bates, C.; Sims-Lucas, S. Endothelial marker-expressing stromal cells are critical for kidney formation. Am. J. Physiol. Renal. Physiol. 2017, 313, F611–F620. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.J.; Lewis, P.; Przepiorski, A.; Sander, V. Turning mesoderm into kidney. Semin. Cell Dev. Biol. 2019, 91, 86–93. [Google Scholar] [CrossRef]
- Jacob, M.; Yusuf, F.; Jacob, H.J. Development, differentiation and derivatives of the Wolffian and Müllerian ducts. In The Human Embryo; Yamada, S., Takakuwa, T., Eds.; IntechOpen: London, UK, 2012. [Google Scholar] [CrossRef]
- Dressler, G.R. Patterning and early cell lineage decisions in the developing kidney: The role of Pax genes. Pediatr. Nephrol. 2011, 26, 1387–1394. [Google Scholar] [CrossRef]
- Lindstrom, N.O.; De Sena Brandine, G.; Tran, T.; Ransick, A.; Suh, G.; Guo, J.; Kim, A.D.; Parvez, R.K.; Ruffins, S.W.; Rutledge, E.A.; et al. Progressive Recruitment of Mesenchymal Progenitors Reveals a Time-Dependent Process of Cell Fate Acquisition in Mouse and Human Nephrogenesis. Dev. Cell 2018, 45, 651–660.e4. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, N.O.; Guo, J.; Kim, A.D.; Tran, T.; Guo, Q.; De Sena Brandine, G.; Ransick, A.; Parvez, R.K.; Thornton, M.E.; Basking, L.; et al. Conserved and Divergent Features of Mesenchymal Progenitor Cell Types within the Cortical Nephrogenic Niche of the Human and Mouse Kidney. J. Am. Soc. Nephrol. 2018, 29, 806–824. [Google Scholar] [CrossRef]
- Cullen-McEwen, L.A.; Sutherland, M.R.; Black, M.J. The Human Kidney: Parallels in Structure, Spatial Development, and Timing of Nephrogenesis. In Kidney Development, Disease, Repair and Regeneration, 1st ed.; Little, M., Ed.; Academic Press: London, UK, 2016; Volume 1, pp. 27–40. [Google Scholar]
- Lin, Y.; Zhang, S.; Tuukkanen, J.; Peltoketo, H.; Pihlajaniemi, T.; Vainio, S. Patterning parameters associated with the branching of the ureteric bud regulated by epithelial-mesenchymal interactions. Int. J. Dev. Biol. 2003, 47, 3–13. [Google Scholar]
- Watanabe, T.; Costantini, F. Real-time analysis of ureteric bud branching morphogenesis in vitro. Dev. Biol. 2004, 271, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Michos, O.; Shakya, R.; Riccio, P.; Enomoto, H.; Licht, J.D.; Asai, N.; Takahashi, M.; Ohgami, N.; Kato, M.; et al. Ret-dependent cell rearrangements in the Wolffian duct epithelium initiate ureteric bud morphogenesis. Dev. Cell 2009, 17, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Costantini, F. Genetic controls and cellular behaviors in branching morphogenesis of the renal collecting system. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 693–713. [Google Scholar] [CrossRef]
- Sariola, H. Nephron induction revisited: From caps to condensates. Curr. Opin. Nephrol. Hypertens. 2002, 11, 17–21. [Google Scholar] [CrossRef]
- Xu, P.X.; Zheng, W.; Huang, L.; Maire, P.; Laclef, C.; Silvius, D. Six1 is required for the early organogenesis of mammalian kidney. Development 2003, 130, 3085–3094. [Google Scholar] [CrossRef]
- Nie, X.; Xu, J.; El-Hashash, A.; Xu, P.X. Six1 regulates Grem1 expression in the metanephric mesenchyme to initiate branching morphogenesis. Dev. Biol. 2011, 352, 141–151. [Google Scholar] [CrossRef]
- Hartman, H.A.; Lai, H.L.; Patterson, L.T. Cessation of renal morphogenesis in mice. Dev. Biol. 2007, 310, 379–387. [Google Scholar] [CrossRef]
- Rumballe, B.A.; Georgas, K.M.; Combes, A.N.; Ju, A.L.; Gilbert, T.; Little, M.H. Nephron formation adopts a novel spatial topology at cessation of nephrogenesis. Dev. Biol. 2011, 360, 110–122. [Google Scholar] [CrossRef]
- Cebrian, C.; Borodo, K.; Charles, N.; Herzlinger, D.A. Morphometric index of the developing murine kidney. Dev. Dyn. 2004, 231, 601–608. [Google Scholar] [CrossRef]
- Short, K.M.; Combes, A.N.; Lefevre, J.; Ju, A.L.; Georgas, K.M.; Lamberton, T.; Cairncross, O.; Rumballe, B.A.; McMahon, A.P.; Hamilton, N.A.; et al. Global quantification of tissue dynamics in the developing mouse kidney. Dev. Cell 2014, 29, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Gomez-Pardo, E.; Dressler, G.R.; Gruss, P. Pax-2 controls multiple steps of urogenital development. Development 1995, 121, 4057–4065. [Google Scholar]
- Kreidberg, J.A.; Sariola, H.; Loring, J.M.; Maeda, M.; Pelletier, J.; Housman, D.; Jaenisch, R. WT-1 is required for early kidney development. Cell 1993, 74, 679–691. [Google Scholar] [CrossRef]
- Davies, J.A.; Ladomery, M.; Hohenstein, P.; Michael, L.; Shafe, A.; Spraggon, L.; Hastie, N. Development of an siRNA-based method for repressing specific genes in renal organ culture and its use to show that the Wt1 tumour suppressor is required for nephron differentiation. Hum. Mol. Genet. 2004, 13, 235–246. [Google Scholar] [CrossRef]
- Xu, P.X.; Adams, J.; Peters, H.; Brown, M.C.; Heaney, S.; Maas, R. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat. Genet. 1999, 23, 113–117. [Google Scholar] [CrossRef]
- Wellik, D.M.; Hawkes, P.J.; Capecchi, M.R. Hox11 paralogous genes are essential for metanephric kidney induction. Genes Dev. 2002, 16, 1423–1432. [Google Scholar] [CrossRef]
- O’Brien, L.L. Nephron progenitor cell commitment: Striking the right balance. Semin. Cell Dev. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Costantini, F.; Kopan, R. Patterning a complex organ: Branching morphogenesis and nephron segmentation in kidney development. Dev. Cell 2010, 18, 698–712. [Google Scholar] [CrossRef]
- Walker, K.A.; Sims-Lucas, S.; Bates, C.M. Fibroblast growth factor receptor signaling in kidney and lower urinary tract development. Pediatr. Nephrol. 2016, 31, 885–895. [Google Scholar] [CrossRef]
- Kurtzeborn, K.; Cebrian, C.; Kuure, S. Regulation of Renal Differentiation by Trophic Factors. Front. Physiol. 2018, 9, 1588. [Google Scholar] [CrossRef]
- Dumbrava, M.G.; Lacanlale, J.L.; Rowan, C.J.; Rosenblum, N.D. Transforming growth factor beta signaling functions during mammalian kidney development. Pediatr. Nephrol. 2020. [Google Scholar] [CrossRef]
- Lu, B.C.; Cebrian, C.; Chi, X.; Kuure, S.; Kuo, R.; Bates, C.M.; Arber, S.; Hassell, J.; MacNeil, L.; Hoshi, M.; et al. Etv4 and Etv5 are required downstream of GDNF and Ret for kidney branching morphogenesis. Nat. Genet. 2009, 41, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Kuure, S.; Chi, X.; Lu, B.; Costantini, F. The transcription factors Etv4 and Etv5 mediate formation of the ureteric bud tip domain during kidney development. Development 2010, 137, 1975–1979. [Google Scholar] [CrossRef]
- Riccio, P.; Cebrian, C.; Zong, H.; Hippenmeyer, S.; Costantini, F. Ret and Etv4 Promote Directed Movements of Progenitor Cells during Renal Branching Morphogenesis. PLoS Biol. 2016, 14, e1002382. [Google Scholar] [CrossRef]
- Sampogna, R.V.; Schneider, L.; Al-Awqati, Q. Developmental Programming of Branching Morphogenesis in the Kidney. J. Am. Soc. Nephrol. 2015, 26, 2414–2422. [Google Scholar] [CrossRef]
- Michael, L.; Davies, J.A. Pattern and regulation of cell proliferation during murine ureteric bud development. J. Anat. 2004, 204, 241–255. [Google Scholar] [CrossRef]
- Packard, A.; Georgas, K.; Michos, O.; Riccio, P.; Cebrian, C.; Combes, A.N.; Ju, A.; Ferrer-Vaquer, A.; Hadjantonakis, A.K.; Zong, H.; et al. Luminal Mitosis Drives Epithelial Cell Dispersal within the Branching Ureteric Bud. Dev. Cell 2013, 27, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Kuure, S.; Cebrian, C.; Machingo, Q.; Lu, B.C.; Chi, X.; Hyink, D.; D’Agati, V.; Gurniak, C.; Witke, W.; Costantini, F. Actin depolymerizing factors cofilin1 and destrin are required for ureteric bud branching morphogenesis. PLoS Genet. 2010, 6, e1001176. [Google Scholar] [CrossRef]
- Elias, B.C.; Das, A.; Parekh, D.V.; Mernaugh, G.; Adams, R.; Yang, Z.; Brakebusch, C.; Pozzi, A.; Marciano, D.K.; Carroll, T.J.; et al. Cdc42 regulates epithelial cell polarity and cytoskeletal function during kidney tubule development. J. Cell Sci. 2015, 128, 4293–4305. [Google Scholar] [CrossRef]
- Marciano, D.K.; Brakeman, P.R.; Lee, C.Z.; Spivak, N.; Eastburn, D.J.; Bryant, D.M.; Beaudoin, G.M., 3rd; Hofmann, I.; Mostov, K.E.; Reichardt, L.F. p120 catenin is required for normal renal tubulogenesis and glomerulogenesis. Development 2011, 138, 2099–2109. [Google Scholar] [CrossRef]
- Karner, C.M.; Chirumamilla, R.; Aoki, S.; Igarashi, P.; Wallingford, J.B.; Carroll, T.J. Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nat. Genet. 2009, 41, 793–799. [Google Scholar] [CrossRef]
- Lienkamp, S.S.; Liu, K.; Karner, C.M.; Carroll, T.J.; Ronneberger, O.; Wallingford, J.B.; Walz, G. Vertebrate kidney tubules elongate using a planar cell polarity-dependent, rosette-based mechanism of convergent extension. Nat. Genet. 2012, 44, 1382–1387. [Google Scholar] [CrossRef] [PubMed]
- Menshykau, D.; Michos, O.; Lang, C.; Conrad, L.; McMahon, A.P.; Iber, D. Image-based modeling of kidney branching morphogenesis reveals GDNF-RET based Turing-type mechanism and pattern-modulating WNT11 feedback. Nat. Commun. 2019, 10, 239. [Google Scholar] [CrossRef]
- Lefevre, J.G.; Short, K.M.; Lamberton, T.O.; Michos, O.; Graf, D.; Smyth, I.M.; Hamilton, N.A. Branching morphogenesis in the developing kidney is governed by rules that pattern the ureteric tree. Development 2017, 144, 4377–4385. [Google Scholar] [CrossRef]
- Little, M.H.; Lawlor, K.T. Recreating, expanding and using nephron progenitor populations. Nat. Rev. Nephrol. 2020, 16, 75–76. [Google Scholar] [CrossRef]
- Levinson, R.; Mendelsohn, C. Stromal progenitors are important for patterning epithelial and mesenchymal cell types in the embryonic kidney. Semin. Cell Dev. Biol. 2003, 14, 225–231. [Google Scholar] [CrossRef]
- Moore, M.W.; Klein, R.D.; Farinas, I.; Sauer, H.; Armanini, M.; Phillips, H.; Reichardt, L.F.; Ryan, A.M.; Carver-Moore, K.; Rosenthal, A. Renal and neuronal abnormalities in mice lacking GDNF. Nature 1996, 382, 76–79. [Google Scholar] [CrossRef]
- Sanchez, M.P.; Silos-Santiago, I.; Frisen, J.; He, B.; Lira, S.A.; Barbacid, M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature 1996, 382, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Sainio, K.; Suvanto, P.; Davies, J.; Wartiovaara, J.; Wartiovaara, K.; Saarma, M.; Arumae, U.; Meng, X.; Lindahl, M.; Pachnis, V.; et al. Glial-cell-line-derived neurotrophic factor is required for bud initiation from ureteric epithelium. Development 1997, 124, 4077–4087. [Google Scholar]
- Hellmich, H.L.; Kos, L.; Cho, E.S.; Mahon, K.A.; Zimmer, A. Embryonic expression of glial cell-line derived neurotrophic factor (GDNF) suggests multiple developmental roles in neural differentiation and epithelial-mesenchymal interactions. Mech. Dev. 1996, 54, 95–105. [Google Scholar] [CrossRef]
- Suvanto, P.; Hiltunen, J.O.; Arumae, U.; Moshnyakov, M.; Sariola, H.; Sainio, K.; Saarma, M. Localization of glial cell line-derived neurotrophic factor (GDNF) mRNA in embryonic rat by in situ hybridization. Eur. J. Neurosci. 1996, 8, 816–822. [Google Scholar] [CrossRef]
- Shakya, R.; Watanabe, T.; Costantini, F. The role of GDNF/Ret signaling in ureteric bud cell fate and branching morphogenesis. Dev. Cell 2005, 8, 65–74. [Google Scholar] [CrossRef]
- Kumar, A.; Kopra, J.; Varendi, K.; Porokuokka, L.L.; Panhelainen, A.; Kuure, S.; Marshall, P.; Karalija, N.; Harma, M.A.; Vilenius, C.; et al. GDNF Overexpression from the Native Locus Reveals its Role in the Nigrostriatal Dopaminergic System Function. PLoS Genet. 2015, 11, e1005710. [Google Scholar] [CrossRef]
- Varendi, K.; Kumar, A.; Harma, M.A.; Andressoo, J.O. miR-1, miR-10b, miR-155, and miR-191 are novel regulators of BDNF. Cell. Mol. Life Sci. 2014, 71, 4443–4456. [Google Scholar] [CrossRef]
- Li, H.; Jakobson, M.; Ola, R.; Gui, Y.; Kumar, A.; Sipila, P.; Sariola, H.; Kuure, S.; Andressoo, J.O. Development of the urogenital system is regulated via the 3′UTR of GDNF. Sci. Rep. 2019, 9, 5302. [Google Scholar] [CrossRef]
- Jain, S.; Encinas, M.; Johnson, E.M., Jr.; Milbrandt, J. Critical and distinct roles for key RET tyrosine docking sites in renal development. Genes Dev. 2006, 20, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Golden, J.P.; Wozniak, D.; Pehek, E.; Johnson, E.M., Jr.; Milbrandt, J. RET is dispensable for maintenance of midbrain dopaminergic neurons in adult mice. J. Neurosci. 2006, 26, 11230–11238. [Google Scholar] [CrossRef]
- Ihermann-Hella, A.; Hirashima, T.; Kupari, J.; Kurtzeborn, K.; Li, H.; Kwon, H.N.; Cebrian, C.; Soofi, A.; Dapkunas, A.; Miinalainen, I.; et al. Dynamic MAPK/ERK Activity Sustains Nephron Progenitors through Niche Regulation and Primes Precursors for Differentiation. Stem Cell Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ihermann-Hella, A.; Lume, M.; Miinalainen, I.J.; Pirttiniemi, A.; Gui, Y.; Peranen, J.; Charron, J.; Saarma, M.; Costantini, F.; Kuure, S. Mitogen-activated protein kinase (MAPK) pathway regulates branching by remodeling epithelial cell adhesion. PLoS Genet. 2014, 10, e1004193. [Google Scholar] [CrossRef]
- Larue, L.; Ohsugi, M.; Hirchenhain, J.; Kemler, R. E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc. Natl. Acad. Sci. USA 1994, 91, 8263–8267. [Google Scholar] [CrossRef]
- Riethmacher, D.; Brinkmann, V.; Birchmeier, C. A targeted mutation in the mouse E-cadherin gene results in defective preimplantation development. Proc. Natl. Acad. Sci. USA 1995, 92, 855–859. [Google Scholar] [CrossRef]
- Chen, H.; Guo, R.; Zhang, Q.; Guo, H.; Yang, M.; Wu, Z.; Gao, S.; Liu, L.; Chen, L. Erk signaling is indispensable for genomic stability and self-renewal of mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E5936–E5943. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, H.; Chen, L. A dual role of Erk signaling in embryonic stem cells. Exp. Hematol. 2016, 44, 151–156. [Google Scholar] [CrossRef][Green Version]
- Blomqvist, S.R.; Vidarsson, H.; Fitzgerald, S.; Johansson, B.R.; Ollerstam, A.; Brown, R.; Persson, A.E.; Bergstrom, G.G.; Enerback, S. Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J. Clin. Invest. 2004, 113, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, L.; Zhou, Q.; Zhang, X.; Berger, S.; Bi, J.; Lewis, D.E.; Xia, Y.; Zhang, W. Aqp2-expressing cells give rise to renal intercalated cells. J. Am. Soc. Nephrol. 2013, 24, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wang, Y.; Tripathi, P.; Manda, K.R.; Mukherjee, M.; Chaklader, M.; Austin, P.F.; Surendran, K.; Chen, F. Adam10 mediates the choice between principal cells and intercalated cells in the kidney. J. Am. Soc. Nephrol. 2015, 26, 149–159. [Google Scholar] [CrossRef]
- Jeong, H.W.; Jeon, U.S.; Koo, B.K.; Kim, W.Y.; Im, S.K.; Shin, J.; Cho, Y.; Kim, J.; Kong, Y.Y. Inactivation of Notch signaling in the renal collecting duct causes nephrogenic diabetes insipidus in mice. J. Clin. Invest. 2009, 119, 3290–3300. [Google Scholar] [CrossRef]
- Grassmeyer, J.; Mukherjee, M.; deRiso, J.; Hettinger, C.; Bailey, M.; Sinha, S.; Visvader, J.E.; Zhao, H.; Fogarty, E.; Surendran, K. Elf5 is a principal cell lineage specific transcription factor in the kidney that contributes to Aqp2 and Avpr2 gene expression. Dev. Biol. 2017, 424, 77–89. [Google Scholar] [CrossRef]
- El-Dahr, S.S.; Li, Y.; Liu, J.; Gutierrez, E.; Hering-Smith, K.S.; Signoretti, S.; Pignon, J.C.; Sinha, S.; Saifudeen, Z. p63+ ureteric bud tip cells are progenitors of intercalated cells. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Werth, M.; Schmidt-Ott, K.M.; Leete, T.; Qiu, A.; Hinze, C.; Viltard, M.; Paragas, N.; Shawber, C.J.; Yu, W.; Lee, P.; et al. Transcription factor TFCP2L1 patterns cells in the mouse kidney collecting ducts. eLife 2017, 6. [Google Scholar] [CrossRef]
- Mukherjee, M.; DeRiso, J.; Janga, M.; Fogarty, E.; Surendran, K. Foxi1 inactivation rescues loss of principal cell fate selection in Hes1-deficient kidneys but does not ensure maintenance of principal cell gene expression. Dev. Biol. 2020, 466, 1–11. [Google Scholar] [CrossRef]
- Mukherjee, M.; Fogarty, E.; Janga, M.; Surendran, K. Notch Signaling in Kidney Development, Maintenance, and Disease. Biomolecules 2019, 9, 692. [Google Scholar] [CrossRef]
- Rao, R.; Bhalla, V.; Pastor-Soler, N.M. Intercalated Cells of the Kidney Collecting Duct in Kidney Physiology. Semin. Nephrol. 2019, 39, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Mugford, J.W.; Krautzberger, A.M.; Naiman, N.; Liao, J.; McMahon, A.P. Identification of a Multipotent Self-Renewing Stromal Progenitor Population during Mammalian Kidney Organogenesis. Stem Cell Rep. 2014, 3, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef]
- Naiman, N.; Fujioka, K.; Fujino, M.; Valerius, M.T.; Potter, S.S.; McMahon, A.P.; Kobayashi, A. Repression of Interstitial Identity in Nephron Progenitor Cells by Pax2 Establishes the Nephron-Interstitium Boundary during Kidney Development. Dev. Cell 2017, 41, 349–365.e3. [Google Scholar] [CrossRef]
- Boyle, S.C.; Liu, Z.Y.; Kopan, R. Notch signaling is required for the formation of mesangial cells from a stromal mesenchyme precursor during kidney development. Development 2014, 141, 346–354. [Google Scholar] [CrossRef]
- Grigorieva, I.V.; Oszwald, A.; Grigorieva, E.F.; Schachner, H.; Neudert, B.; Ostendorf, T.; Floege, J.; Lindenmeyer, M.T.; Cohen, C.D.; Panzer, U.; et al. A Novel Role for GATA3 in Mesangial Cells in Glomerular Development and Injury. J. Am. Soc. Nephrol. 2019, 30, 1641–1658. [Google Scholar] [CrossRef] [PubMed]
- England, A.R.; Chaney, C.P.; Das, A.; Patel, M.; Malewska, A.; Armendariz, D.; Hon, G.C.; Strand, D.W.; Drake, K.A.; Carroll, T.J. Identification and characterization of cellular heterogeneity within the developing renal interstitium. Development 2020, 147. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Tanigawa, S.; Karner, C.M.; Xin, M.; Lum, L.; Chen, C.; Olson, E.N.; Perantoni, A.O.; Carroll, T.J. Stromal-epithelial crosstalk regulates kidney progenitor cell differentiation. Nat. Cell Biol. 2013, 15, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Bagherie-Lachidan, M.; Reginensi, A.; Pan, Q.; Zaveri, H.P.; Scott, D.A.; Blencowe, B.J.; Helmbacher, F.; McNeill, H. Stromal Fat4 acts non-autonomously with Dchs1/2 to restrict the nephron progenitor pool. Development 2015, 142, 2564–2573. [Google Scholar] [CrossRef]
- Mao, Y.; Francis-West, P.; Irvine, K.D. Fat4/Dchs1 signaling between stromal and cap mesenchyme cells influences nephrogenesis and ureteric bud branching. Development 2015, 142, 2574–2585. [Google Scholar] [CrossRef]
- Ohmori, T.; Tanigawa, S.; Kaku, Y.; Fujimura, S.; Nishinakamura, R. Sall1 in renal stromal progenitors non-cell autonomously restricts the excessive expansion of nephron progenitors. Sci. Rep. 2015, 5, 15676. [Google Scholar] [CrossRef]
- Mugford, J.W.; Sipila, P.; Kobayashi, A.; Behringer, R.R.; McMahon, A.P. Hoxd11 specifies a program of metanephric kidney development within the intermediate mesoderm of the mouse embryo. Dev. Biol. 2008, 319, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Patterson, L.T.; Pembaur, M.; Potter, S.S. Hoxa11 and Hoxd11 regulate branching morphogenesis of the ureteric bud in the developing kidney. Development 2001, 128, 2153–2161. [Google Scholar]
- Miyazaki, Y.; Oshima, K.; Fogo, A.; Hogan, B.L.; Ichikawa, I. Bone morphogenetic protein 4 regulates the budding site and elongation of the mouse ureter. J. Clin. Invest. 2000, 105, 863–873. [Google Scholar] [CrossRef]
- Grieshammer, U.; Le, M.; Plump, A.S.; Wang, F.; Tessier-Lavigne, M.; Martin, G.R. SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Dev. Cell 2004, 6, 709–717. [Google Scholar] [CrossRef]
- Wainwright, E.N.; Wilhelm, D.; Combes, A.N.; Little, M.H.; Koopman, P. ROBO2 restricts the nephrogenic field and regulates Wolffian duct-nephrogenic cord separation. Dev. Biol. 2015, 404, 88–102. [Google Scholar] [CrossRef]
- Michos, O.; Goncalves, A.; Lopez-Rios, J.; Tiecke, E.; Naillat, F.; Beier, K.; Galli, A.; Vainio, S.; Zeller, R. Reduction of BMP4 activity by gremlin 1 enables ureteric bud outgrowth and GDNF/WNT11 feedback signalling during kidney branching morphogenesis. Development 2007, 134, 2397–2405. [Google Scholar] [CrossRef]
- Basson, M.A.; Akbulut, S.; Watson-Johnson, J.; Simon, R.; Carroll, T.J.; Shakya, R.; Gross, I.; Martin, G.R.; Lufkin, T.; McMahon, A.P.; et al. Sprouty1 is a critical regulator of GDNF/RET-mediated kidney induction. Dev. Cell 2005, 8, 229–239. [Google Scholar] [CrossRef]
- Kuure, S.; Vuolteenaho, R.; Vainio, S. Kidney morphogenesis: Cellular and molecular regulation. Mech. Dev. 2000, 92, 31–45. [Google Scholar] [CrossRef]
- Lindstrom, N.O.; Carragher, N.O.; Hohenstein, P. The PI3K pathway balances self-renewal and differentiation of nephron progenitor cells through beta-catenin signaling. Stem Cell Rep. 2015, 4, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Combes, A.N.; Lefevre, J.G.; Wilson, S.; Hamilton, N.A.; Little, M.H. Cap mesenchyme cell swarming during kidney development is influenced by attraction, repulsion, and adhesion to the ureteric tip. Dev. Biol. 2016, 418, 297–306. [Google Scholar] [CrossRef]
- Muller, U.; Wang, D.; Denda, S.; Meneses, J.J.; Pedersen, R.A.; Reichardt, L.F. Integrin alpha8beta1 is critically important for epithelial-mesenchymal interactions during kidney morphogenesis. Cell 1997, 88, 603–613. [Google Scholar] [CrossRef]
- Brandenberger, R.; Schmidt, A.; Linton, J.; Wang, D.; Backus, C.; Denda, S.; Muller, U.; Reichardt, L.F. Identification and characterization of a novel extracellular matrix protein nephronectin that is associated with integrin alpha8beta1 in the embryonic kidney. J. Cell Biol. 2001, 154, 447–458. [Google Scholar] [CrossRef]
- Shaw, L.; Johnson, P.A.; Kimber, S.J. Gene expression profiling of the developing mouse kidney and embryo. In Vitro Cell. Dev. Biol. Anim. 2010, 46, 155–165. [Google Scholar] [CrossRef]
- Uchiyama, Y.; Sakaguchi, M.; Terabayashi, T.; Inenaga, T.; Inoue, S.; Kobayashi, C.; Oshima, N.; Kiyonari, H.; Nakagata, N.; Sato, Y.; et al. Kif26b, a kinesin family gene, regulates adhesion of the embryonic kidney mesenchyme. Proc. Natl. Acad. Sci. USA 2010, 107, 9240–9245. [Google Scholar] [CrossRef]
- Chen, S.; Brunskill, E.W.; Potter, S.S.; Dexheimer, P.J.; Salomonis, N.; Aronow, B.J.; Hong, C.I.; Zhang, T.; Kopan, R. Intrinsic Age-Dependent Changes and Cell-Cell Contacts Regulate Nephron Progenitor Lifespan. Dev. Cell 2015, 35, 49–62. [Google Scholar] [CrossRef]
- O’Brien, L.L.; Combes, A.N.; Short, K.M.; Lindstrom, N.O.; Whitney, P.H.; Cullen-McEwen, L.A.; Ju, A.; Abdelhalim, A.; Michos, O.; Bertram, J.F.; et al. Wnt11 directs nephron progenitor polarity and motile behavior ultimately determining nephron endowment. eLife 2018, 7. [Google Scholar] [CrossRef]
- Brown, A.C.; Muthukrishnan, S.D.; Guay, J.A.; Adams, D.C.; Schafer, D.A.; Fetting, J.L.; Oxburgh, L. Role for compartmentalization in nephron progenitor differentiation. Proc. Natl. Acad. Sci. USA 2013, 110, 4640–4645. [Google Scholar] [CrossRef]
- Boyle, S.; Misfeldt, A.; Chandler, K.J.; Deal, K.K.; Southard-Smith, E.M.; Mortlock, D.P.; Baldwin, H.S.; de Caestecker, M. Fate mapping using Cited1-CreERT2 mice demonstrates that the cap mesenchyme contains self-renewing progenitor cells and gives rise exclusively to nephronic epithelia. Dev. Biol. 2008, 313, 234–245. [Google Scholar] [CrossRef]
- Combes, A.N.; Wilson, S.; Phipson, B.; Binnie, B.B.; Ju, A.; Lawlor, K.T.; Cebrian, C.; Walton, S.L.; Smyth, I.M.; Moritz, K.M.; et al. Haploinsufficiency for the Six2 gene increases nephron progenitor proliferation promoting branching and nephron number. Kidney Int. 2018, 93, 589–598. [Google Scholar] [CrossRef]
- Mugford, J.W.; Yu, J.; Kobayashi, A.; McMahon, A.P. High-resolution gene expression analysis of the developing mouse kidney defines novel cellular compartments within the nephron progenitor population. Dev. Biol. 2009, 333, 312–323. [Google Scholar] [CrossRef]
- Self, M.; Lagutin, O.V.; Bowling, B.; Hendrix, J.; Cai, Y.; Dressler, G.R.; Oliver, G. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 2006, 25, 5214–5228. [Google Scholar] [CrossRef]
- Boyle, S.; Shioda, T.; Perantoni, A.O.; de Caestecker, M. Cited1 and Cited2 are differentially expressed in the developing kidney but are not required for nephrogenesis. Dev. Dyn. 2007, 236, 2321–2330. [Google Scholar] [CrossRef]
- Lawlor, K.T.; Zappia, L.; Lefevre, J.; Park, J.S.; Hamilton, N.A.; Oshlack, A.; Little, M.H.; Combes, A.N. Nephron progenitor commitment is a stochastic process influenced by cell migration. eLife 2019, 8. [Google Scholar] [CrossRef]
- Park, J.S.; Ma, W.; O’Brien, L.L.; Chung, E.; Guo, J.J.; Cheng, J.G.; Valerius, M.T.; McMahon, J.A.; Wong, W.H.; McMahon, A.P. Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev. Cell 2012, 23, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.; Sutherland, M.R.; Flores, T.J.; Kent, A.L.; Dahlstrom, J.E.; Puelles, V.G.; Bertram, J.F.; McMahon, A.P.; Little, M.H.; Moore, L.; et al. Development of the Human Fetal Kidney from Mid to Late Gestation in Male and Female Infants. EBioMedicine 2018, 27, 275–283. [Google Scholar] [CrossRef]
- Brown, A.C.; Muthukrishnan, S.D.; Oxburgh, L. A synthetic niche for nephron progenitor cells. Dev. Cell 2015, 34, 229–241. [Google Scholar] [CrossRef]
- Ramalingam, H.; Fessler, A.R.; Das, A.; Valerius, M.T.; Basta, J.; Robbins, L.; Brown, A.C.; Oxburgh, L.; McMahon, A.P.; Rauchman, M.; et al. Disparate levels of beta-catenin activity determine nephron progenitor cell fate. Dev. Biol. 2018, 440, 13–21. [Google Scholar] [CrossRef]
- Saifudeen, Z. Tissue-Specific Functions of p53 During Kidney Development. Results Probl. Cell Differ. 2017, 60, 111–136. [Google Scholar] [CrossRef] [PubMed]
- Ferre, S.; Igarashi, P. New insights into the role of HNF-1beta in kidney (patho)physiology. Pediatr. Nephrol. 2019, 34, 1325–1335. [Google Scholar] [CrossRef]
- El-Dahr, S.S.; Saifudeen, Z. Epigenetic regulation of renal development. Semin. Cell Dev. Biol. 2019, 91, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Little, M.H.; Combes, A.N. Kidney organoids: Accurate models or fortunate accidents. Genes Dev. 2019, 33, 1319–1345. [Google Scholar] [CrossRef]
- Oxburgh, L.; Muthukrishnan, S.D.; Brown, A. Growth Factor Regulation in the Nephrogenic Zone of the Developing Kidney. Results Probl. Cell Differ. 2017, 60, 137–164. [Google Scholar] [CrossRef] [PubMed]
- Saxen, L.; Lehtonen, E. Transfilter induction of kidney tubules as a function of the extent and duration of intercellular contacts. J. Embryol. Exp. Morphol. 1978, 47, 97–109. [Google Scholar]
- Barasch, J.; Yang, J.; Ware, C.B.; Taga, T.; Yoshida, K.; Erdjument-Bromage, H.; Tempst, P.; Parravicini, E.; Malach, S.; Aranoff, T.; et al. Mesenchymal to epithelial conversion in rat metanephros is induced by LIF. Cell 1999, 99, 377–386. [Google Scholar] [CrossRef]
- Schmidt-Ott, K.M.; Masckauchan, T.N.; Chen, X.; Hirsh, B.J.; Sarkar, A.; Yang, J.; Paragas, N.; Wallace, V.A.; Dufort, D.; Pavlidis, P.; et al. beta-catenin/TCF/Lef controls a differentiation-associated transcriptional program in renal epithelial progenitors. Development 2007, 134, 3177–3190. [Google Scholar] [CrossRef] [PubMed]
- Kuure, S.; Popsueva, A.; Jakobson, M.; Sainio, K.; Sariola, H. Glycogen synthase kinase-3 inactivation and stabilization of beta-catenin induce nephron differentiation in isolated mouse and rat kidney mesenchymes. J. Am. Soc. Nephrol. 2007, 18, 1130–1139. [Google Scholar] [CrossRef]
- Stark, K.; Vainio, S.; Vassileva, G.; McMahon, A.P. Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 1994, 372, 679–683. [Google Scholar] [CrossRef]
- Carroll, T.J.; Park, J.S.; Hayashi, S.; Majumdar, A.; McMahon, A.P. Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the Mammalian urogenital system. Dev. Cell 2005, 9, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Boyle, S.C.; Kim, M.; Valerius, M.T.; McMahon, A.P.; Kopan, R. Notch pathway activation can replace the requirement for Wnt4 and Wnt9b in mesenchymal-to-epithelial transition of nephron stem cells. Development 2011, 138, 4245–4254. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Valerius, M.T.; McMahon, A.P. Wnt/beta-catenin signaling regulates nephron induction during mouse kidney development. Development 2007, 134, 2533–2539. [Google Scholar] [CrossRef]
- Pan, X.; Karner, C.M.; Carroll, T.J. Myc cooperates with beta-catenin to drive gene expression in nephron progenitor cells. Development 2017, 144, 4173–4182. [Google Scholar] [CrossRef]
- Karner, C.M.; Das, A.; Ma, Z.; Self, M.; Chen, C.; Lum, L.; Oliver, G.; Carroll, T.J. Canonical Wnt9b signaling balances progenitor cell expansion and differentiation during kidney development. Development 2011, 138, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, S.M.; Robbins, L.; Rauchman, M. Conditional expression of Wnt9b in Six2-positive cells disrupts stomach and kidney function. PLoS ONE 2012, 7, e43098. [Google Scholar] [CrossRef] [PubMed]
- Vidal, V.P.; Jian-Motamedi, F.; Rekima, S.; Gregoire, E.P.; Szenker-Ravi, E.; Leushacke, M.; Reversade, B.; Chaboissier, M.C.; Schedl, A. R-spondin signalling is essential for the maintenance and differentiation of mouse nephron progenitors. eLife 2020, 9. [Google Scholar] [CrossRef]
- Burn, S.F.; Webb, A.; Berry, R.L.; Davies, J.A.; Ferrer-Vaquer, A.; Hadjantonakis, A.K.; Hastie, N.D.; Hohenstein, P. Calcium/NFAT signalling promotes early nephrogenesis. Dev. Biol. 2011, 352, 288–298. [Google Scholar] [CrossRef]
- Gilbert, T.; Leclerc, C.; Moreau, M. Control of kidney development by calcium ions. Biochimie 2011, 93, 2126–2131. [Google Scholar] [CrossRef]
- Tanigawa, S.; Wang, H.; Yang, Y.; Sharma, N.; Tarasova, N.; Ajima, R.; Yamaguchi, T.P.; Rodriguez, L.G.; Perantoni, A.O. Wnt4 induces nephronic tubules in metanephric mesenchyme by a non-canonical mechanism. Dev. Biol. 2011, 352, 58–69. [Google Scholar] [CrossRef]
- Poladia, D.P.; Kish, K.; Kutay, B.; Hains, D.; Kegg, H.; Zhao, H.; Bates, C.M. Role of fibroblast growth factor receptors 1 and 2 in the metanephric mesenchyme. Dev. Biol. 2006, 291, 325–339. [Google Scholar] [CrossRef]
- Brown, A.C.; Adams, D.; de Caestecker, M.; Yang, X.; Friesel, R.; Oxburgh, L. FGF/EGF signaling regulates the renewal of early nephron progenitors during embryonic development. Development 2011, 138, 5099–5112. [Google Scholar] [CrossRef] [PubMed]
- Barak, H.; Huh, S.H.; Chen, S.; Jeanpierre, C.; Martinovic, J.; Parisot, M.; Bole-Feysot, C.; Nitschke, P.; Salomon, R.; Antignac, C.; et al. FGF9 and FGF20 maintain the stemness of nephron progenitors in mice and man. Dev. Cell 2012, 22, 1191–1207. [Google Scholar] [CrossRef]
- Zhou, M.; Sutliff, R.L.; Paul, R.J.; Lorenz, J.N.; Hoying, J.B.; Haudenschild, C.C.; Yin, M.; Coffin, J.D.; Kong, L.; Kranias, E.G.; et al. Fibroblast growth factor 2 control of vascular tone. Nat. Med. 1998, 4, 201–207. [Google Scholar] [CrossRef]
- Miller, D.L.; Ortega, S.; Bashayan, O.; Basch, R.; Basilico, C. Compensation by fibroblast growth factor 1 (FGF1) does not account for the mild phenotypic defects observed in FGF2 null mice. Mol. Cell. Biol. 2000, 20, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Huh, S.H.; Ha, L.; Jang, H.S. Nephron Progenitor Maintenance Is Controlled through Fibroblast Growth Factors and Sprouty1 Interaction. J. Am. Soc. Nephrol. 2020, 31, 2559–2572. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.; Zhang, S.; Lin, Y.; Prunskaite-Hyyrylainen, R.; Vuolteenaho, R.; Itaranta, P.; Vainio, S. Sprouty proteins regulate ureteric branching by coordinating reciprocal epithelial Wnt11, mesenchymal Gdnf and stromal Fgf7 signalling during kidney development. Development 2004, 131, 3345–3356. [Google Scholar] [CrossRef]
- Rozen, E.J.; Schmidt, H.; Dolcet, X.; Basson, M.A.; Jain, S.; Encinas, M. Loss of Sprouty1 rescues renal agenesis caused by Ret mutation. J. Am. Soc. Nephrol. 2009, 20, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Michos, O.; Cebrian, C.; Hyink, D.; Grieshammer, U.; Williams, L.; D’Agati, V.; Licht, J.D.; Martin, G.R.; Costantini, F. Kidney development in the absence of Gdnf and Spry1 requires Fgf10. PLoS Genet. 2010, 6, e1000809. [Google Scholar] [CrossRef]
- Hida, M.; Omori, S.; Awazu, M. ERK and p38 MAP kinase are required for rat renal development. Kidney Int. 2002, 61, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Osafune, K.; Takasato, M.; Kispert, A.; Asashima, M.; Nishinakamura, R. Identification of multipotent progenitors in the embryonic mouse kidney by a novel colony-forming assay. Development 2006, 133, 151–161. [Google Scholar] [CrossRef]
- Blank, U.; Brown, A.; Adams, D.C.; Karolak, M.J.; Oxburgh, L. BMP7 promotes proliferation of nephron progenitor cells via a JNK-dependent mechanism. Development 2009, 136, 3557–3566. [Google Scholar] [CrossRef]
- Nakane, A.; Kojima, Y.; Hayashi, Y.; Kohri, K.; Masui, S.; Nishinakamura, R. Pax2 overexpression in embryoid bodies induces upregulation of integrin alpha8 and aquaporin-1. In Vitro Cell. Dev. Biol. Anim. 2009, 45, 62–68. [Google Scholar] [CrossRef]
- Muthukrishnan, S.D.; Yang, X.; Friesel, R.; Oxburgh, L. Concurrent BMP7 and FGF9 signalling governs AP-1 function to promote self-renewal of nephron progenitor cells. Nat. Commun. 2015, 6, 10027. [Google Scholar] [CrossRef]
- Togel, F.; Valerius, M.T.; Freedman, B.S.; Iatrino, R.; Grinstein, M.; Bonventre, J.V. Repair after nephron ablation reveals limitations of neonatal neonephrogenesis. JCI Insight 2017, 2, e88848. [Google Scholar] [CrossRef]
- Volovelsky, O.; Nguyen, T.; Jarmas, A.E.; Combes, A.N.; Wilson, S.B.; Little, M.H.; Witte, D.P.; Brunskill, E.W.; Kopan, R. Hamartin regulates cessation of mouse nephrogenesis independently of Mtor. Proc. Natl. Acad. Sci. USA 2018, 115, 5998–6003. [Google Scholar] [CrossRef]
- Yermalovich, A.V.; Osborne, J.K.; Sousa, P.; Han, A.; Kinney, M.A.; Chen, M.J.; Robinton, D.A.; Montie, H.; Pearson, D.S.; Wilson, S.B.; et al. Lin28 and let-7 regulate the timing of cessation of murine nephrogenesis. Nat. Commun. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Urbach, A.; Yermalovich, A.; Zhang, J.; Spina, C.S.; Zhu, H.; Perez-Atayde, A.R.; Shukrun, R.; Charlton, J.; Sebire, N.; Mifsud, W.; et al. Lin28 sustains early renal progenitors and induces Wilms tumor. Genes Dev. 2014, 28, 971–982. [Google Scholar] [CrossRef]
- Li, Z.; Araoka, T.; Wu, J.; Liao, H.K.; Li, M.; Lazo, M.; Zhou, B.; Sui, Y.; Wu, M.Z.; Tamura, I.; et al. 3D Culture Supports Long-Term Expansion of Mouse and Human Nephrogenic Progenitors. Cell Stem Cell 2016. [Google Scholar] [CrossRef]
- Morizane, R.; Bonventre, J.V. Generation of nephron progenitor cells and kidney organoids from human pluripotent stem cells. Nat. Protoc. 2017, 12, 195–207. [Google Scholar] [CrossRef]
- Tanigawa, S.; Taguchi, A.; Sharma, N.; Perantoni, A.O.; Nishinakamura, R. Selective In Vitro Propagation of Nephron Progenitors Derived from Embryos and Pluripotent Stem Cells. Cell Rep. 2016, 15, 801–813. [Google Scholar] [CrossRef]
- Chen, K.S.; Stroup, E.K.; Budhipramono, A.; Rakheja, D.; Nichols-Vinueza, D.; Xu, L.; Stuart, S.H.; Shukla, A.A.; Fraire, C.; Mendell, J.T.; et al. Mutations in microRNA processing genes in Wilms tumors derepress the IGF2 regulator PLAG1. Genes Dev. 2018, 32, 996–1007. [Google Scholar] [CrossRef]
- Ogawa, O.; Eccles, M.R.; Szeto, J.; McNoe, L.A.; Yun, K.; Maw, M.A.; Smith, P.J.; Reeve, A.E. Relaxation of insulin-like growth factor II gene imprinting implicated in Wilms’ tumour. Nature 1993, 362, 749–751. [Google Scholar] [CrossRef]
- Treger, T.D.; Chowdhury, T.; Pritchard-Jones, K.; Behjati, S. The genetic changes of Wilms tumour. Nat. Rev. Nephrol. 2019, 15, 240–251. [Google Scholar] [CrossRef]
- Wegert, J.; Ishaque, N.; Vardapour, R.; Georg, C.; Gu, Z.; Bieg, M.; Ziegler, B.; Bausenwein, S.; Nourkami, N.; Ludwig, N.; et al. Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell 2015, 27, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Ruf, R.G.; Xu, P.X.; Silvius, D.; Otto, E.A.; Beekmann, F.; Muerb, U.T.; Kumar, S.; Neuhaus, T.J.; Kemper, M.J.; Raymond, R.M., Jr.; et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc. Natl. Acad. Sci. USA 2004, 101, 8090–8095. [Google Scholar] [CrossRef]
- Kusafuka, T.; Miao, J.; Kuroda, S.; Udatsu, Y.; Yoneda, A. Codon 45 of the beta-catenin gene, a specific mutational target site of Wilms’ tumor. Int. J. Mol. Med. 2002, 10, 395–399. [Google Scholar]
- Beckwith, J.B.; Kiviat, N.B.; Bonadio, J.F. Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms’ tumor. Pediatr. Pathol. 1990, 10, 1–36. [Google Scholar] [CrossRef]
- Dekel, B.; Metsuyanim, S.; Schmidt-Ott, K.M.; Fridman, E.; Jacob-Hirsch, J.; Simon, A.; Pinthus, J.; Mor, Y.; Barasch, J.; Amariglio, N.; et al. Multiple imprinted and stemness genes provide a link between normal and tumor progenitor cells of the developing human kidney. Cancer Res. 2006, 66, 6040–6049. [Google Scholar] [CrossRef]
- Gadd, S.; Huff, V.; Huang, C.C.; Ruteshouser, E.C.; Dome, J.S.; Grundy, P.E.; Breslow, N.; Jennings, L.; Green, D.M.; Beckwith, J.B.; et al. Clinically relevant subsets identified by gene expression patterns support a revised ontogenic model of Wilms tumor: A Children’s Oncology Group Study. Neoplasia 2012, 14, 742–756. [Google Scholar] [CrossRef]
- Berry, R.L.; Ozdemir, D.D.; Aronow, B.; Lindstrom, N.O.; Dudnakova, T.; Thornburn, A.; Perry, P.; Baldock, R.; Armit, C.; Joshi, A.; et al. Deducing the stage of origin of Wilms’ tumours from a developmental series of Wt1-mutant mice. Dis. Models Mech. 2015, 8, 903–917. [Google Scholar] [CrossRef]
- Pode-Shakked, N.; Shukrun, R.; Mark-Danieli, M.; Tsvetkov, P.; Bahar, S.; Pri-Chen, S.; Goldstein, R.S.; Rom-Gross, E.; Mor, Y.; Fridman, E.; et al. The isolation and characterization of renal cancer initiating cells from human Wilms’ tumour xenografts unveils new therapeutic targets. EMBO Mol. Med. 2013, 5, 18–37. [Google Scholar] [CrossRef]
- Shukrun, R.; Pode-Shakked, N.; Pleniceanu, O.; Omer, D.; Vax, E.; Peer, E.; Pri-Chen, S.; Jacob, J.; Hu, Q.; Harari-Steinberg, O.; et al. Wilms’ tumor blastemal stem cells dedifferentiate to propagate the tumor bulk. Stem Cell Rep. 2014, 3, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Schutgens, F.; Rookmaaker, M.B.; Margaritis, T.; Rios, A.; Ammerlaan, C.; Jansen, J.; Gijzen, L.; Vormann, M.; Vonk, A.; Viveen, M.; et al. Tubuloids derived from human adult kidney and urine for personalized disease modeling. Nat. Biotechnol. 2019, 37, 303–313. [Google Scholar] [CrossRef]
- Fukuzawa, R.; Anaka, M.R.; Morison, I.M.; Reeve, A.E. The developmental programme for genesis of the entire kidney is recapitulated in Wilms tumour. PLoS ONE 2017, 12, e0186333. [Google Scholar] [CrossRef]
- Young, M.D.; Mitchell, T.J.; Vieira Braga, F.A.; Tran, M.G.B.; Stewart, B.J.; Ferdinand, J.R.; Collord, G.; Botting, R.A.; Popescu, D.M.; Loudon, K.W.; et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 2018, 361, 594–599. [Google Scholar] [CrossRef]
- Drake, K.A.; Chaney, C.P.; Das, A.; Roy, P.; Kwartler, C.S.; Rakheja, D.; Carroll, T.J. Stromal beta-catenin activation impacts nephron progenitor differentiation in the developing kidney and may contribute to Wilms tumor. Development 2020, 147. [Google Scholar] [CrossRef]
- Ollila, S.; Domenech-Moreno, E.; Laajanen, K.; Wong, I.P.L.; Tripathi, S.; Pentinmikko, N.; Gao, Y.J.; Yan, Y.; Niemela, E.H.; Wang, T.C.; et al. Stromal Lkb1 deficiency leads to gastrointestinal tumorigenesis involving the IL-11-JAK/STAT3 pathway. J. Clin. Investig. 2018, 128, 402–414. [Google Scholar] [CrossRef]
- Hastie, N.D. The genetics of Wilms’ tumor—A case of disrupted development. Annu. Rev. Genet. 1994, 28, 523–558. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Hohenstein, P.; Kuure, S. Embryonic Kidney Development, Stem Cells and the Origin of Wilms Tumor. Genes 2021, 12, 318. https://doi.org/10.3390/genes12020318
Li H, Hohenstein P, Kuure S. Embryonic Kidney Development, Stem Cells and the Origin of Wilms Tumor. Genes. 2021; 12(2):318. https://doi.org/10.3390/genes12020318
Chicago/Turabian StyleLi, Hao, Peter Hohenstein, and Satu Kuure. 2021. "Embryonic Kidney Development, Stem Cells and the Origin of Wilms Tumor" Genes 12, no. 2: 318. https://doi.org/10.3390/genes12020318
APA StyleLi, H., Hohenstein, P., & Kuure, S. (2021). Embryonic Kidney Development, Stem Cells and the Origin of Wilms Tumor. Genes, 12(2), 318. https://doi.org/10.3390/genes12020318