
Precision Surgery and Kidney Cancer: Knowledge of Genetic Alterations Influences Surgical Management


Abstract
1. Introduction
2. Von Hippel-Lindau (VHL)
3. Birt-Hogg-Dubé (BHD)
4. Hereditary Papillary Renal Carcinoma (HPRC)
5. Hereditary Leiomyomatosis Renal Cell Cancer (HLRCC)
6. Succinate Dehydrogenase (SDH)-Deficient RCC
7. BAP1-Tumor Predisposition Syndrome (BAP1-TPS)
8. Tuberous Sclerosis Complex (TSC)
9. Clinical Management
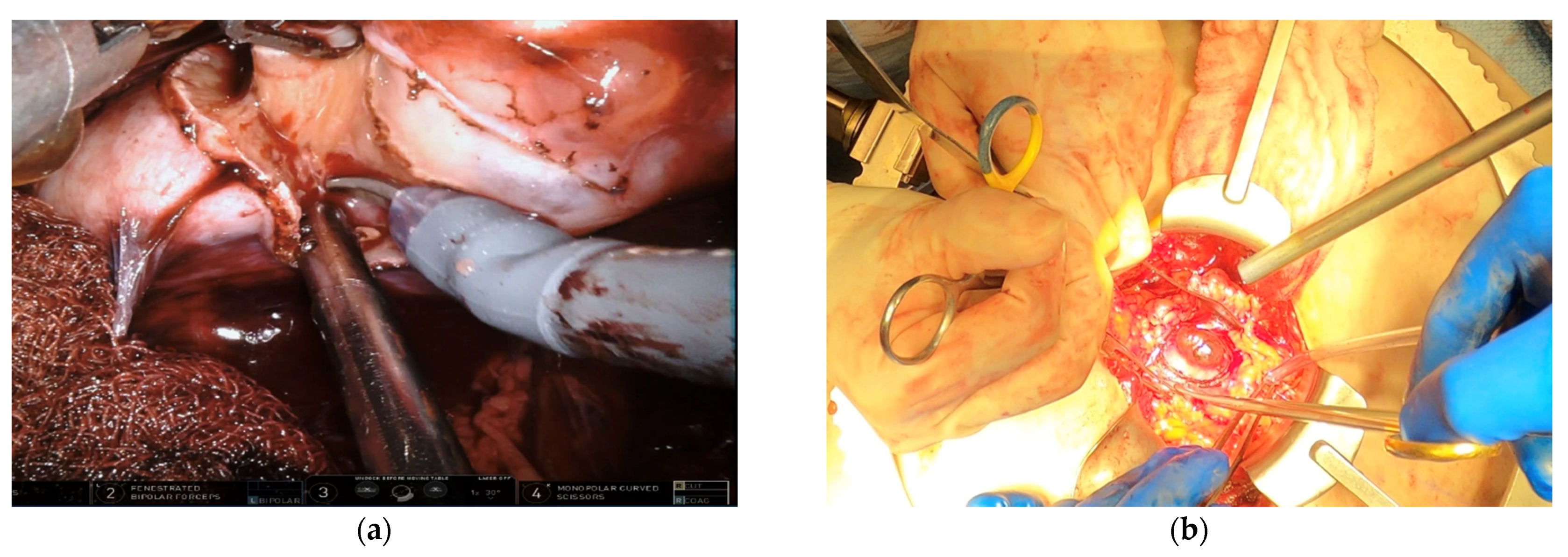
10. Precision Surgery: When to Intervene
11. Precision Surgery: How to Intervene
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Miller, K.D.; Siegel, R.L.; Khan, R.; Jemal, A. Cancer Statistics. Cancer Rehabil. 2018, 70, 7–30. [Google Scholar] [CrossRef]
- Ball, M.W.; Shuch, B.M. Inherited kidney cancer syndromes. Curr. Opin. Urol. 2019, 29, 334–343. [Google Scholar] [CrossRef]
- Ricketts, C.J.; Vocke, C.D.; Lang, M.; Chen, X.; Zhao, Y.; Tran, B.; Tandon, M.; Schmidt, L.S.; Ball, M.W.; Linehan, W.M. A germline 1;3 translocation disrupting the VHL gene: A novel genetic cause for von Hippel-Lindau. J. Med. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Vocke, C.D.; Ricketts, C.J.; Metwalli, A.R.; Ball, M.W.; Schmidt, L.S.; Linehan, W.M. Clinical and Molecular Characterization of Microphthalmia-Associated Transcription Factor (MITF)-Related Renal Cell Carcinoma. Urology 2020. [Google Scholar] [CrossRef] [PubMed]
- Vocke, C.D.; Ricketts, C.J.; Ball, M.W.; Schmidt, L.S.; Metwalli, A.R.; Middelton, L.A.; Killian, J.K.; Khan, J.; Meltzer, P.S.; Simonds, W.F.; et al. CDC73 Germline Mutation in a Family with Mixed Epithelial and Stromal Tumors. Urology 2019, 124, 91–97. [Google Scholar] [CrossRef]
- Linehan, W.M.; Schmidt, L.S.; Crooks, D.R.; Wei, D.; Srinivasan, R.; Lang, M.; Ricketts, C.J. The Metabolic Basis of Kidney Cancer. Cancer Discov. 2019, 9, 1006–1021. [Google Scholar] [CrossRef]
- Haas, N.B.; Nathanson, K.L. Hereditary Kidney Cancer Syndromes. Adv. Chronic Kidney Dis. 2014, 21, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.H.; Graff, R.E.; Holst, K.K.; Moeller, S.; Unger, R.H.; et al. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 2016, 315, 68–76. [Google Scholar] [CrossRef]
- Von Hippel, E. Die anatomische Grundlage der von mir beschriebenen „sehr seltenen Erkrankung der Netzhaut. Albrecht Graefes Arch. Ophthalmol. 1911, 79, 350–377. [Google Scholar] [CrossRef]
- Lindau, A. Zur frage der angiomatosis retinae und ihrer hirnkomplikationen. Acta Ophthalmol. 2009, 4, 193–226. [Google Scholar] [CrossRef]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Genetics of human cancer. Ann. Rev. Genet. 1986, 20, 231–251. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFα Targeted for VHL-Mediated Destruction by Proline Hydroxylation: Implications for O2 Sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Kaelin, W.G. Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. von Hippel-Lindau disease. Lancet 2003, 361, 2059–2067. [Google Scholar] [CrossRef]
- Hes, F.; Zewald, R.; Peeters, T.; Sijmons, R.; Links, T.; Verheij, J.; Matthijs, G.; Legius, E.; Mortier, G.; Van Der Torren, K.; et al. Genotype-phenotype correlations in families with deletions in the von Hippel-Lindau (VHL) gene. Qual. Life Res. 2000, 106, 425–431. [Google Scholar] [CrossRef]
- Maher, E.R.; Webster, A.R.; Richards, F.M.; Green, J.S.; A Crossey, P.; Payne, S.J.; Moore, A.T. Phenotypic expression in von Hippel-Lindau disease: Correlations with germline VHL gene mutations. J. Med. Genet. 1996, 33, 328–332. [Google Scholar] [CrossRef][Green Version]
- Shuch, B.; Vourganti, S.; Ricketts, C.J.; Middleton, L.; Peterson, J.; Merino, M.J.; Metwalli, A.R.; Srinivasan, R.; Linehan, W.M. Defining Early-Onset Kidney Cancer: Implications for Germline and Somatic Mutation Testing and Clinical Management. J. Clin. Oncol. 2014, 32, 431–437. [Google Scholar] [CrossRef]
- Schmidt, L.S.; Warren, M.B.; Nickerson, M.L.; Weirich, G.; Matrosova, V.; Toro, J.R.; Turner, M.L.; Duray, P.; Merino, M.; Hewitt, S.; et al. Birt-Hogg-Dubé Syndrome, a Genodermatosis Associated with Spontaneous Pneumothorax and Kidney Neoplasia, Maps to Chromosome 17p11.2. Am. J. Hum. Genet. 2001, 69, 876–882. [Google Scholar] [CrossRef]
- Nickerson, M.L.; Warren, M.B.; Toro, J.R.; Matrosova, V.; Glenn, G.; Turner, M.L.; Duray, P.; Merino, M.; Choyke, P.; Pavlovich, C.P.; et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell 2002, 2, 157–164. [Google Scholar] [CrossRef]
- Schmidt, L.S. Birt-Hogg-Dubé syndrome: From gene discovery to molecularly targeted therapies. Fam. Cancer 2012, 12, 357–364. [Google Scholar] [CrossRef]
- Pavlovich, C.P.; Grubb, R.L., 3rd; Hurley, K.; Glenn, G.M.; Toro, J.; Schmidt, L.S.; Torres-Cabala, C.; Merino, M.J.; Zbar, B.; Choyke, P.; et al. Evaluation and management of renal tumors in the Birt-Hogg-Dubé syndrome. J. Urol. 2005, 173, 1482–1486. [Google Scholar] [CrossRef]
- Pavlovich, C.P.; Walther, M.M.; Eyler, R.A.; Hewitt, S.M.; Zbar, B.; Linehan, W.M.; Merino, M.J. Renal tumors in the Birt-Hogg-Dubé syndrome. Am. J. Surg. Pathol. 2002, 26, 1542–1552. [Google Scholar] [CrossRef]
- Zbar, B.; Glenn, G.; Lubensky, I.; Choyke, P.; Walther, M.M.; Magnusson, G.; Bergerheim, U.S.; Pettersson, S.; Amin, M.; Hurley, K.; et al. Original Articles: Kidney Cancer: Hereditary Papillary Renal Cell Carcinoma: Clinical Studies in 10 Families. J. Urol. 1995, 153, 907–912. [Google Scholar] [CrossRef]
- Schmidt, L.; Duh, F.-M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Pathirage, G.D.; Alessio, G.; Donald, P.B. Hereditary Papillary Renal Carcinoma Type, I. Curr. Mol. Med. 2004, 4, 855–868. [Google Scholar]
- Schmidt, L.S.; Nickerson, M.L.; Angeloni, D.; Glenn, G.M.; Walther, M.M.; Albert, P.S.; Warren, M.B.; Choyke, P.L.; Torres-Cabala, C.A.; Merino, M.J.; et al. Early onset hereditary papillary renal carcinoma: Germline missense mutations in the tyrosine kinase domain of the met proto-oncogene. J. Urol. 2004, 172, 1256–1261. [Google Scholar] [CrossRef]
- Kloepfer, H.W.; Krafchuk, J.; Derbes, V.; Burks, J. Hereditary Multiple Leiomyoma of the Skin. Am. J. Hum. Genet. 1958, 10, 48–52. [Google Scholar]
- Reed, W.B.; Walker, R.; Horowitz, R. Cutaneous leiomyomata with uterine leiomyomata. Acta Derm. Venereol. 1973, 53, 409–416. [Google Scholar]
- Launonen, V.; Vierimaa, O.; Kiuru, M.; Isola, J.; Roth, S.; Pukkala, E.; Sistonen, P.; Herva, R.; Aaltonen, L.A. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 3387–3392. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.P.M.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.M.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.-L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Grubb, R.L., 3rd; Franks, M.E.; Toro, J.; Middelton, L.; Choyke, L.; Fowler, S.; Torres-Cabala, C.; Glenn, G.M.; Choyke, P.; Merino, M.J.; et al. Hereditary leiomyomatosis and renal cell cancer: A syndrome associated with an aggressive form of inherited renal cancer. J. Urol. 2007, 177, 2074–2079. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.J.; Amin, M.; Smith, S.; Trpkov, K. Chapter 1: Succinate dehydrogenase (SDH) deficient renal carcinoma. In WHO Classification of Tumours of the Urinary System and Male Genital Organs, 4th ed.; Moch, H., Humphrey, P.A., Ulbright, T.M., Reuter, V.E., Eds.; IARC: Lyon, France, 2016; pp. 35–36. [Google Scholar]
- Bardella, C.; Pollard, P.J.; Tomlinson, I. SDH mutations in cancer. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 1432–1443. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Vanharanta, S.; Buchta, M.; McWhinney, S.R.; Virta, S.K.; Peçzkowska, M.; Morrison, C.D.; Lehtonen, R.; Januszewicz, A.; Järvinen, H.; Juhola, M.; et al. Early-Onset Renal Cell Carcinoma as a Novel Extraparaganglial Component of SDHB-Associated Heritable Paraganglioma. Am. J. Hum. Genet. 2004, 74, 153–159. [Google Scholar] [CrossRef]
- Ricketts, C.J.; Shuch, B.; Vocke, C.D.; Metwalli, A.R.; Bratslavsky, G.; Middelton, L.; Yang, Y.; Wei, M.-H.; Pautler, S.E.; Peterson, J.; et al. Succinate Dehydrogenase Kidney Cancer: An Aggressive Example of the Warburg Effect in Cancer. J. Urol. 2012, 188, 2063–2071. [Google Scholar] [CrossRef]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca (2+) flux to mitochondria suppressing cell trans-formation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef]
- Yu, H.; Mashtalir, N.; Daou, S.; Hammond-Martel, I.; Ross, J.; Sui, G.; Hart, G.W.; Rauscher, F.J.; Drobetsky, E.; Milot, E.; et al. The Ubiquitin Carboxyl Hydrolase BAP1 Forms a Ternary Complex with YY1 and HCF-1 and Is a Critical Regulator of Gene Expression. Mol. Cell. Biol. 2010, 30, 5071–5085. [Google Scholar] [CrossRef] [PubMed]
- Popova, T.; Hebert, L.; Jacquemin, V.; Gad, S.; Caux-Moncoutier, V.; Dubois-D’Enghien, C.; Richaudeau, B.; Renaudin, X.; Sellers, J.; Nicolas, A.; et al. Germline BAP1 Mutations Predispose to Renal Cell Carcinomas. Am. J. Hum. Genet. 2013, 92, 974–980. [Google Scholar] [CrossRef]
- Farley, M.N.; Schmidt, L.S.; Mester, J.L.; Pena-Llopis, S.; Pavia-Jimenez, A.; Christie, A.; Vocke, C.D.; Ricketts, C.J.; Peterson, J.; Middeltone, L.; et al. A Novel Germline Mutation in BAP1 Predisposes to Familial Clear-Cell Renal Cell Carcinoma. Mol. Cancer Res. 2013, 11, 1061–1071. [Google Scholar] [CrossRef]
- Rai, K.R.; Pilarski, R.; Cebulla, C.M.; Abdelrahman, M.H. Comprehensive review ofBAP1tumor predisposition syndrome with report of two new cases. Clin. Genet. 2016, 89, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Flores, E.G.; Emi, M.; Johnson, T.A.; Tsunoda, T.; Behner, D.; Hoffman, H.; Hesdorffer, M.; Nasu, M.; Napolitano, A.; et al. Combined Genetic and Genealogic Studies Uncover a Large BAP1 Cancer Syndrome Kindred Tracing Back Nine Generations to a Common Ancestor from the 1700s. PLoS Genet. 2015, 11, e1005633. [Google Scholar] [CrossRef]
- Joseph, R.W.; Kapur, P.; Serie, D.J.; Parasramka, M.; Ho, T.H.; Cheville, J.C.; Frenkel, E.; Parker, A.S.; Brugarolas, J. Clear Cell Renal Cell Carcinoma Subtypes Identified by BAP1 and PBRM1 Expression. J. Urol. 2016, 195, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Bourneville, D.J.A.N. Sclerose tubereuse der circonvolutions cerebrales: Idiotie et epilepsie hemiplegique. Arch de Neurologie. 1880, 1, 81–91. [Google Scholar]
- European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993, 75, 1305–1315. [Google Scholar] [CrossRef]
- Van Slegtenhorst, M.; De Hoogt, R.; Hermans, C.; Nellist, M.; Janssen, B.; Verhoef, S.; Lindhout, D.; Ouweland, A.V.D.; Halley, D.; Young, J.; et al. Identification of the Tuberous Sclerosis Gene TSC1 on Chromosome 9q34. Science 1997, 277, 805–808. [Google Scholar] [CrossRef]
- Van Slegtenhorst, M.; Nellist, M.; Nagelkerken, B.; Cheadle, J.; Snell, R.; van den Ouweland, A.; Reuser, A.; Sampson, J.; Halley, D.; van der Sluijs, P. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 1998, 7, 1053–1057. [Google Scholar] [CrossRef]
- Crino, P.B.; Nathanson, K.L.; Henske, E.P. The Tuberous Sclerosis Complex. N. Engl. J. Med. 2006, 355, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tretiakova, M.S.; Troxell, M.L.; Osunkoya, A.O.; Fadare, O.; Sangoi, A.R.; Shen, S.S.; Lopez-Beltran, A.; Mehra, R.; Heider, A.; et al. Tuberous sclerosis-associated renal cell carcinoma: A clinicopathologic study of 57 separate carcinomas in 18 patients. Am. J. Surg. Pathol. 2014, 38, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Cornejo, K.M.; Sadow, P.M.; Cheng, L.; Wang, M.; Xiao, Y.; Jiang, Z.; Oliva, E.; Jozwiak, S.; Nussbaum, R.L.; et al. Renal Cell Carcinoma in Tuberous Sclerosis Complex. Am. J. Surg. Pathol. 2014, 38, 895–909. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.W.; An, J.Y.; Gomella, P.T.; Gautam, R.; Ricketts, C.J.; Vocke, C.D.; Schmidt, L.S.; Merino, M.J.; Srinivasan, R.; Malayeri, A.A.; et al. Growth Rates of Genetically Defined Renal Tumors: Implications for Active Surveillance and Intervention. J. Clin. Oncol. 2020, 38, 1146–1153. [Google Scholar] [CrossRef]
- Herring, J.C.; Enquist, E.G.; Chernoff, A.; Linehan, W.M.; Choyke, P.L.; Walther, M.M. Parenchymal sparing surgery in patients with hereditary renal cell carcinoma: 10-year experience. J. Urol. 2001, 165, 777–781. [Google Scholar] [CrossRef]
- Campbell, S.; Uzzo, R.G.; Allaf, M.E.; Bass, E.B.; Cadeddu, J.A.; Chang, A.; Clark, P.E.; Davis, B.J.; Derweesh, I.H.; Giambarresi, L.; et al. Renal Mass and Localized Renal Cancer: AUA Guideline. J. Urol. 2017, 198, 520–529. [Google Scholar] [CrossRef]
- Hemal, A.K.; Menon, M. Robotics in Genitourinary Surgery; Springer: Berlin, Germany, 2018. [Google Scholar]
- Blom, J.H.; Van Poppel, H.; Maréchal, J.M.; Jacqmin, D.; Schröder, F.H.; De Prijck, L.; Sylvester, R. Radical Nephrectomy with and without Lymph-Node Dissection: Final Results of European Organization for Research and Treatment of Cancer (EORTC) Randomized Phase 3 Trial 30881. Eur. Urol. 2009, 55, 28–34. [Google Scholar] [CrossRef]


{kind=link}
| Germline Alteration | BHD | VHL | BAP1 | HLRCC |
|---|---|---|---|---|
 |  |  |  | |
| Tumor size | 2.5 cm | 2.5 cm | 2.5 cm | 2 cm |
| Active Surveillance versus Surgery | Active surveillance | Active surveillance | Surgery or Active Surveillance | Immediate Surgery |
| Next Imaging Follow-up | 24 months * | 12 months * | 6 months if surgery deferred | Immediate Surgery |
| Surgical Approach | Robotic surgery | Robotic surgery | Robotic surgery | Strongly consider open surgery |
| Extent of Resection | Tumor enucleation | Tumor enucleation | Partial nephrectomy with margin | Partial nephrectomy with wide margin or radical nephrectomy. Consider regional lymph node dissection, especially for larger and centrally located tumors |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomella, P.T.; Linehan, W.M.; Ball, M.W. Precision Surgery and Kidney Cancer: Knowledge of Genetic Alterations Influences Surgical Management. Genes 2021, 12, 261. https://doi.org/10.3390/genes12020261
Gomella PT, Linehan WM, Ball MW. Precision Surgery and Kidney Cancer: Knowledge of Genetic Alterations Influences Surgical Management. Genes. 2021; 12(2):261. https://doi.org/10.3390/genes12020261
Chicago/Turabian StyleGomella, Patrick T., W. Marston Linehan, and Mark W. Ball. 2021. "Precision Surgery and Kidney Cancer: Knowledge of Genetic Alterations Influences Surgical Management" Genes 12, no. 2: 261. https://doi.org/10.3390/genes12020261
APA StyleGomella, P. T., Linehan, W. M., & Ball, M. W. (2021). Precision Surgery and Kidney Cancer: Knowledge of Genetic Alterations Influences Surgical Management. Genes, 12(2), 261. https://doi.org/10.3390/genes12020261