Characterization and Comparative Analysis of Complete Mitogenomes of Three Cacatua Parrots (Psittaciformes: Cacatuidae)


Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics and Sampling
2.2. DNA Extraction and Molecular Identification
2.3. Library Preparation and Next Generation Sequencing
2.4. Mitochondrial Genome Analysis
2.5. Control Region Analysis
2.6. Phylogenetic Analysis and Molecular Dating
3. Results and Discussion
3.1. Mitogenome Structure and Composition
3.2. Protein Coding Genes
3.3. Ribosomal and Transfer RNA Genes
3.4. Degenerated Genes
3.5. Control Regions
3.6. Gene Arrangements
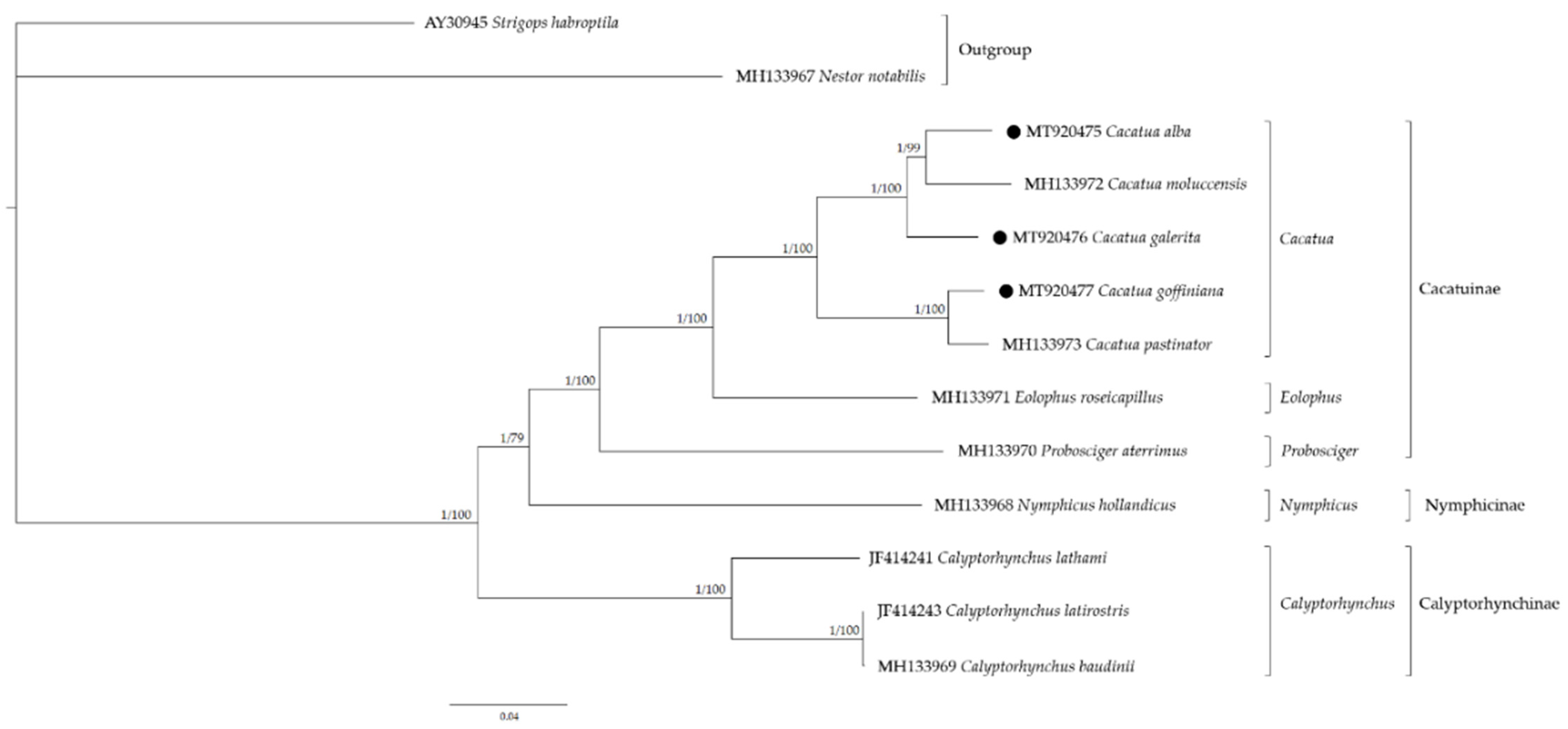
3.7. Phylogenetic Analysis and Molecular Dating
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cameron, M. Cockatoos; CSIRO PUBLISHING: Collingwood, Australia, 2007; pp. 12–113. [Google Scholar]
- del Hoyo, J. All the Birds of the World; Lynx Edicions: Barcelona, Spain, 2020. [Google Scholar]
- Shepherd, C.R.; Sukumaran, J.; Wich, S.A. Open Season: An Analysis of the Pet Trade in Medan, Sumatra, 1997–2001; TRAFFIC Southeast Asia: Selangor, Malaysia, 2004; pp. 8–36. [Google Scholar]
- The IUCN Red List OF Threatened Species. Available online: https://www.iucnredlist.org (accessed on 26 October 2020).
- Gibb, G.C.; Kardailsky, O.; Kimball, R.T.; Braun, E.L.; Penny, D. Mitochondrial Genomes and Avian Phylogeny: Complex Characters and Resolvability without Explosive Radiations. Mol. Biol. Evol. 2007, 24, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Dimitriou, A.C.; Forcina, G.; Papazoglou, C.; Panayides, P.; Guerrini, M.; Crabtree, A.; Barbanera, F.; Sfenthourakis, S. DNA Barcoding of Bird Species in Cyprus: A Tool for Conservation Purposes. Bird Conserv. Int. 2017, 27, 483. [Google Scholar] [CrossRef]
- Moritz, C. Applications of Mitochondrial DNA Analysis in Conservation: A Critical Review. Mol. Ecol. 1994, 3, 401–411. [Google Scholar] [CrossRef]
- Hebert, P.D.; Cywinska, A.; Ball, S.L.; Dewaard, J.R. Biological Identifications through DNA Barcodes. Proc. Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Tsai, L.; Huang, M.; Jhuang, J.; Yao, C.; Chin, S.; Wang, L.; Linacre, A.; Hsieh, H. A Novel Strategy for Avian Species Identification by Cytochrome B Gene. Electrophoresis 2008, 29, 2413–2418. [Google Scholar] [CrossRef]
- Coghlan, M.L.; White, N.E.; Parkinson, L.; Haile, J.; Spencer, P.B.; Bunce, M. Egg Forensics: An Appraisal of DNA Sequencing to Assist in Species Identification of Illegally Smuggled Eggs. Forensic Sci. Int. Genet. 2012, 6, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, P.F.; Oliveira-Marques, A.R.; Matsumoto, T.E.; Miyaki, C.Y. DNA Barcoding Identifies Illegal Parrot Trade. J. Hered. 2015, 106, 560–564. [Google Scholar] [CrossRef]
- Wright, T.F.; Wilkinson, G.S. Population Genetic Structure and Vocal Dialects in an Amazon Parrot. Proc. Biol. Sci. 2001, 268, 609–616. [Google Scholar] [CrossRef]
- Faria, P.J.; Guedes, N.M.; Yamashita, C.; Martuscelli, P.; Miyaki, C.Y. Genetic Variation and Population Structure of the Endangered Hyacinth Macaw (Anodorhynchus Hyacinthinus): Implications for Conservation. Biodivers. Conserv. 2008, 17, 765–779. [Google Scholar] [CrossRef]
- Haghani, A.; Aliabadian, M.; Sarhangzadeh, J.; Setoodeh, A. Evaluation of Genetic Diversity and Population Structure of Macqueen’s Bustard Chlamydotis Macqueenii in Iran. Bird Study 2018, 65, 108–113. [Google Scholar] [CrossRef]
- Varela, A.I.; Brokordt, K.; Ismar-Rebitz, S.M.; Gaskin, C.P.; Carlile, N.; O’Dwyer, T.; Adams, J.; VanderWerf, E.A.; Luna-Jorquera, G. Genetic Diversity, Population Structure, and Historical Demography of a Highly Vagile and Human-impacted Seabird in the Pacific Ocean: The Red-tailed Tropicbird. Phaethon Rubricauda. Aquat. Conserv. 2020, 1–11. [Google Scholar] [CrossRef]
- Boore, J.L. Animal Mitochondrial Genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, J.R.; Wright, T.F.; Bermingham, E. Duplication and Concerted Evolution of the Mitochondrial Control Region in the Parrot Genus Amazona. Mol. Biol. Evol. 2001, 18, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Schirtzinger, E.E.; Tavares, E.S.; Gonzales, L.A.; Eberhard, J.R.; Miyaki, C.Y.; Sanchez, J.J.; Hernandez, A.; Müeller, H.; Graves, G.R.; Fleischer, R.C. Multiple Independent Origins of Mitochondrial Control Region Duplications in the Order Psittaciformes. Mol. Phylogenet. Evol. 2012, 64, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, J.R.; Wright, T.F. Rearrangement and Evolution of Mitochondrial Genomes in Parrots. Mol. Phylogenet. Evol. 2016, 94, 34–46. [Google Scholar] [CrossRef]
- Urantówka, A.D.; Kroczak, A.; Silva, T.; Padrón, R.Z.; Gallardo, N.F.; Blanch, J.; Blanch, B.; Mackiewicz, P. New Insight into Parrots’ Mitogenomes Indicates that their Ancestor Contained a Duplicated Region. Mol. Biol. Evol. 2018, 35, 2989–3009. [Google Scholar] [CrossRef]
- Clayton, D.A. Replication and Transcription of Vertebrate Mitochondrial DNA. Annu. Rev. Cell Biol. 1991, 7, 453–478. [Google Scholar] [CrossRef]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA Maintenance in Vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef]
- Sbisà, E.; Tanzariello, F.; Reyes, A.; Pesole, G.; Saccone, C. Mammalian Mitochondrial D-Loop Region Structural Analysis: Identification of New Conserved Sequences and their Functional and Evolutionary Implications. Gene 1997, 205, 125–140. [Google Scholar] [CrossRef]
- Ruokonen, M.; Kvist, L. Structure and Evolution of the Avian Mitochondrial Control Region. Mol. Phylogenet. Evol. 2002, 23, 422–432. [Google Scholar] [CrossRef]
- Walberg, M.W.; Clayton, D.A. Sequence and Properties of the Human KB Cell and Mouse L Cell D-Loop Regions of Mitochondrial DNA. Nucleic Acids Res. 1981, 9, 5411–5421. [Google Scholar] [CrossRef] [PubMed]
- White, N.E.; Phillips, M.J.; Gilbert, M.T.P.; Alfaro-Núñez, A.; Willerslev, E.; Mawson, P.R.; Spencer, P.B.; Bunce, M. The Evolutionary History of Cockatoos (Aves: Psittaciformes: Cacatuidae). Mol. Phylogenet. Evol. 2011, 59, 615–622. [Google Scholar] [CrossRef]
- Wink, M.; Sauer-Gürth, H. Advances in the molecular systematics of African raptors. In Raptors at Risk; Chancellor, R.D., Meyburg, B.-U., Eds.; WWGBP/Handcock House: Berlin, Germany, 2000; pp. 135–147. [Google Scholar]
- Fritz, U.; Auer, M.; Bertolero, A.; Cheylan, M.; Fattizzo, T.; Hundsdörfer, A.K.; Martín Sampayo, M.; Pretus, J.L.; ŠIrokÝ, P.; Wink, M. A Rangewide Phylogeography of Hermann’s Tortoise, Testudo Hermanni (Reptilia: Testudines: Testudinidae): Implications for Taxonomy. Zool. Scr. 2006, 35, 531–543. [Google Scholar] [CrossRef]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing Mitochondrial Genomes Directly from Genomic Next-Generation Sequencing Reads—a Baiting and Iterative Mapping Approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved De Novo Metazoan Mitochondrial Genome Annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN: A Program to Detect tRNA Genes in Metazoan Mitochondrial Nucleotide Sequences. Bioinformatics 2007, 24, 172–175. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of Nucleotide Composition at Fourfold Degenerate Sites of Animal Mitochondrial Genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Lima, N.C.B.; Soares, A.E.R.; Almeida, L.G.; Costa, I.R.; Sato, F.M.; Schneider, P.; Aleixo, A.; Schneider, M.P.; Santos, F.R.; Mello, C.V. Comparative Mitogenomic Analyses of Amazona Parrots and Psittaciformes. Genet. Mol. 2018, 41, 593–604. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of Phylogenies After Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901. [Google Scholar] [CrossRef]
- FigTree v1.4.3. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 29 October 2020).
- Urantówka, A.D.; Kroczak, A.; Mackiewicz, P. The Complete Mitochondrial Genome of Red-Fronted Parrot (Poicephalus gulielmi) Revealed a New Gene Rearrangement within the Order Psittaciformes. Mitochondrial DNA Part B 2017, 2, 833–835. [Google Scholar] [CrossRef]
- Harrison, G.L.; McLenachan, P.A.; Phillips, M.J.; Slack, K.E.; Cooper, A.; Penny, D. Four New Avian Mitochondrial Genomes Help Get to Basic Evolutionary Questions in the Late Cretaceous. Mol. Biol. Evol. 2004, 21, 974–983. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef]
- Mindell, D.P.; Sorenson, M.D.; Dimcheff, D.E. An Extra Nucleotide is Not Translated in Mitochondrial ND3 of some Birds and Turtles. Mol. Biol. Evol. 1998, 15, 1568–1571. [Google Scholar] [CrossRef]
- Kumazawa, Y.; Ota, H.; Nishida, M.; Ozawa, T. Gene Rearrangements in Snake Mitochondrial Genomes: Highly Concerted Evolution of Control-Region-Like Sequences Duplicated and Inserted into a tRNA Gene Cluster. Mol. Biol. Evol. 1996, 13, 1242–1254. [Google Scholar] [CrossRef] [PubMed]
- Umeda, S.; Tang, Y.; Okamoto, M.; Hamasaki, N.; Schon, E.A.; Kang, D. Both Heavy Strand Replication Origins are Active in Partially Duplicated Human Mitochondrial DNAs. Biochem. Biophys. Res. Commun. 2001, 286, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.D.; Good, R.T.; Coleman, R.A.; Lancaster, M.L.; Weeks, A.R. Microsatellite Loci and the Complete Mitochondrial DNA Sequence Characterized through Next Generation Sequencing and De Novo Genome Assembly for the Critically Endangered Orange-Bellied Parrot, Neophema chrysogaster. Mol. Biol. Rep. 2013, 40, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Urantowka, A.D.; Kroczak, A.; Mackiewicz, P. Complete Mitochondrial Genome of the Greater Antillean Parrot Amazona Ventralis (Hispaniolan Amazon). Mitochondrial DNA Part B 2016, 1, 864–866. [Google Scholar] [CrossRef]
- L’abbé, D.; Duhaime, J.; Lang, B.F.; Morais, R. The Transcription of DNA in Chicken Mitochondria Initiates from One Major Bidirectional Promoter. J. Biol. Chem. 1991, 266, 10844–10850. [Google Scholar] [CrossRef]
- Cho, H.; Eda, M.; Nishida, S.; Yasukochi, Y.; Chong, J.; Koike, H. Tandem Duplication of Mitochondrial DNA in the Black-Faced Spoonbill, Platalea Minor. Genes Genet. Syst. 2009, 84, 297–305. [Google Scholar] [CrossRef]
- Morris-Pocock, J.A.; Taylor, S.A.; Birt, T.P.; Friesen, V.L. Concerted Evolution of Duplicated Mitochondrial Control Regions in Three Related Seabird Species. BMC Evol. Biol. 2010, 10, 14. [Google Scholar] [CrossRef]
- Eda, M.; Kuro-o, M.; Higuchi, H.; Hasegawa, H.; Koike, H. Mosaic Gene Conversion After a Tandem Duplication of mtDNA Sequence in Diomedeidae (Albatrosses). Genes Genet. Syst. 2010, 85, 129–139. [Google Scholar] [CrossRef]
- Verkuil, Y.I.; Piersma, T.; Baker, A.J. A Novel Mitochondrial Gene Order in Shorebirds (Scolopacidae, Charadriiformes). Mol. Phylogenet. Evol. 2010, 57, 411–416. [Google Scholar] [CrossRef]
- Sammler, S.; Bleidorn, C.; Tiedemann, R. Full Mitochondrial Genome Sequences of Two Endemic Philippine Hornbill Species (Aves: Bucerotidae) Provide Evidence for Pervasive Mitochondrial DNA Recombination. BMC Genomics. 2011, 12, 35. [Google Scholar] [CrossRef]
- Gibb, G.C.; Kennedy, M.; Penny, D. Beyond Phylogeny: Pelecaniform and Ciconiiform Birds, and Long-Term Niche Stability. Mol. Phylogenet. Evol. 2013, 68, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Lin, Q.; Fang, W.; Chen, X. The Complete Mitochondrial Genomes of Sixteen Ardeid Birds Revealing the Evolutionary Process of the Gene Rearrangements. BMC Genomics 2014, 15, 573. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, Y.; Liu, N.; Yang, J.; Lei, F. Seven Complete Mitochondrial Genome Sequences of Bushtits (Passeriformes, Aegithalidae, Aegithalos): The Evolution Pattern in Duplicated Control Regions. Mitochondrial DNA 2015, 26, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Nishida, C.; Momose, K.; Onuma, M.; Takami, K.; Masuda, R. Gene Duplication and Concerted Evolution of Mitochondrial DNA in Crane Species. Mol. Phylogenet. Evol. 2017, 106, 158–163. [Google Scholar] [CrossRef]
- Mackiewicz, P.; Urantówka, A.D.; Kroczak, A.; Mackiewicz, D. Resolving Phylogenetic Relationships within Passeriformes Based on Mitochondrial Genes and Inferring the Evolution of their Mitogenomes in Terms of Duplications. Genome Biol. Evol. 2019, 11, 2824–2849. [Google Scholar] [CrossRef] [PubMed]
- Ericson, P.G.; Anderson, C.L.; Britton, T.; Elzanowski, A.; Johansson, U.S.; Källersjö, M.; Ohlson, J.I.; Parsons, T.J.; Zuccon, D.; Mayr, G. Diversification of Neoaves: Integration of Molecular Sequence Data and Fossils. Biol. Lett. 2006, 2, 543–547. [Google Scholar] [CrossRef]
- Brown, J.W.; Payne, R.B.; Mindell, D.P. Nuclear DNA does Not Reconcile ‘rocks’ and ‘clocks’ in Neoaves: A Comment on Ericson Et Al. Biol. Lett. 2007, 3, 257–260. [Google Scholar] [CrossRef][Green Version]
- Gelabert, P.; Sandoval-Velasco, M.; Serres, A.; de Manuel, M.; Renom, P.; Margaryan, A.; Stiller, J.; de-Dios, T.; Fang, Q.; Feng, S. Evolutionary History, Genomic Adaptation to Toxic Diet, and Extinction of the Carolina Parakeet. Curr. Biol. 2020, 30, 108–114.e5. [Google Scholar] [CrossRef]
- Boles, W.E. A New Cockatoo (Psittaciformes: Cacatuidae) from the Tertiary of Riversleigh, Northwestern Queensland, and an Evaluation of Rostral Characters in the Systematics of Parrots. Ibis 1993, 135, 8–18. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Subfamily | Species Name | Accession number | References |
|---|---|---|---|---|
| Cacatuidae | Cacatuinae | C. alba | MT920475 | This study |
| C.galerita | MT920476 | This study | ||
| C. goffiniana | MT920477 | This study | ||
| C. moluccensis | MH133972 | [20] | ||
| C. pastinator | MH133973 | [20] | ||
| Eolophus roseicapillus | MH133971 | [20] | ||
| Probosciger aterrimus | MH133970 | [20] | ||
| Calyptorhynchinae | Calyptorhynchus baudinii | MH133969 | [20] | |
| Calyptorhynchus lathami | JF414241 | [26] | ||
| Calyptorhynchus latirostris | JF424243 | [26] | ||
| Nymphicinae | Nymphicus hollandicus | MH133968 | [20] | |
| Psittacidae | Poicephalus gulielmi | MF977813 | [42] | |
| Psittacus erithacus | KM611474 | [19] | ||
| Strigopidae | Nestor notabilis | MH133967 | [20] | |
| Strigops habroptila | AY309456 | [43] |
| Nucleotide Sequence | C. alba | C. galerita | C. goffiniana | |
|---|---|---|---|---|
| Whole sequence | Length (bp) | 18,894 | 18,900 | 19,084 |
| A (%) | 29.5 | 29.4 | 29.7 | |
| C (%) | 31.6 | 31.8 | 31.3 | |
| G (%) | 15.0 | 15.1 | 14.8 | |
| T (%) | 24.0 | 23.7 | 24.2 | |
| A + T (%) | 53.5 | 53.1 | 53.9 | |
| Protein coding genes | Length (bp) | 11,402 | 11,399 | 11,402 |
| A (%) | 29.1 | 29.1 | 29.5 | |
| C (%) | 34.0 | 34.2 | 34.0 | |
| G (%) | 12.9 | 13.0 | 12.6 | |
| T (%) | 23.9 | 23.6 | 24.0 | |
| A + T (%) | 53.0 | 52.7 | 53.5 | |
| Ribosomal RNA genes | Length (bp) | 2543 | 2542 | 2537 |
| A (%) | 32.8 | 32.3 | 33.1 | |
| C (%) | 29.8 | 29.7 | 29.3 | |
| G (%) | 18.9 | 19.3 | 18.7 | |
| T (%) | 18.6 | 18.7 | 18.9 | |
| A + T (%) | 51.4 | 51.0 | 52.0 | |
| Transfer RNA genes | Length (bp) | 1669 | 1673 | 1672 |
| A (%) | 32.5 | 32.4 | 33.1 | |
| C (%) | 25.5 | 26.0 | 25.5 | |
| G (%) | 17.1 | 17.1 | 16.5 | |
| T (%) | 24.7 | 24.5 | 24.9 | |
| A + T (%) | 57.3 | 56.9 | 58.0 | |
| Control regions | Length (bp) | 2428 | 2430 | 2591 |
| A (%) | 22.6 | 22.6 | 21.9 | |
| C (%) | 25.8 | 26.1 | 24.7 | |
| G (%) | 18.9 | 19.1 | 19.3 | |
| T (%) | 32.8 | 32.2 | 34.0 | |
| A + T (%) | 55.4 | 54.8 | 55.9 | |
| Species | cytb | nd6 | tRNA-Glu | References | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Functional gene (bp) | Degenerated gene (bp) | Sequence Similarity (%) | Functional gene (bp) | Degenerated gene (bp) | Sequence Similarity (%) | Functional gene (bp) | Degenerated gene (bp) | Sequence Similarity (%) | ||
| C. alba | 1140 | 114 | 92.1 | 519 | 650 | 92.9 | 75 | 50 | 50.0 | This study |
| C. galerita | 1140 | 115 | 88.7 | 519 | 653 | 92.4 | 75 | 50 | 41.8 | This study |
| C. goffiniana | 1140 | 113 | 94.6 | 519 | 680 | 90.8 | 75 | 51 | 41.5 | This study |
| C. moluccensis | 1140 | 115 | 92.2 | 507 | 644 | 95.0 | 75 | 35 | 60.6 | [20] |
| C. pastinator * | 1140 | 115 | 99.1 | 519 | 519 | 100 | 75 | 75 | 100 | [20] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-I.; Do, T.D.; Choi, Y.; Yeo, Y.; Kim, C.-B. Characterization and Comparative Analysis of Complete Mitogenomes of Three Cacatua Parrots (Psittaciformes: Cacatuidae). Genes 2021, 12, 209. https://doi.org/10.3390/genes12020209
Kim J-I, Do TD, Choi Y, Yeo Y, Kim C-B. Characterization and Comparative Analysis of Complete Mitogenomes of Three Cacatua Parrots (Psittaciformes: Cacatuidae). Genes. 2021; 12(2):209. https://doi.org/10.3390/genes12020209
Chicago/Turabian StyleKim, Jung-Il, Thinh Dinh Do, Yisoo Choi, Yonggu Yeo, and Chang-Bae Kim. 2021. "Characterization and Comparative Analysis of Complete Mitogenomes of Three Cacatua Parrots (Psittaciformes: Cacatuidae)" Genes 12, no. 2: 209. https://doi.org/10.3390/genes12020209
APA StyleKim, J.-I., Do, T. D., Choi, Y., Yeo, Y., & Kim, C.-B. (2021). Characterization and Comparative Analysis of Complete Mitogenomes of Three Cacatua Parrots (Psittaciformes: Cacatuidae). Genes, 12(2), 209. https://doi.org/10.3390/genes12020209