Integrated Analysis of Methylomic and Transcriptomic Data to Identify Potential Diagnostic Biomarkers for Major Depressive Disorder


Abstract
1. Introduction
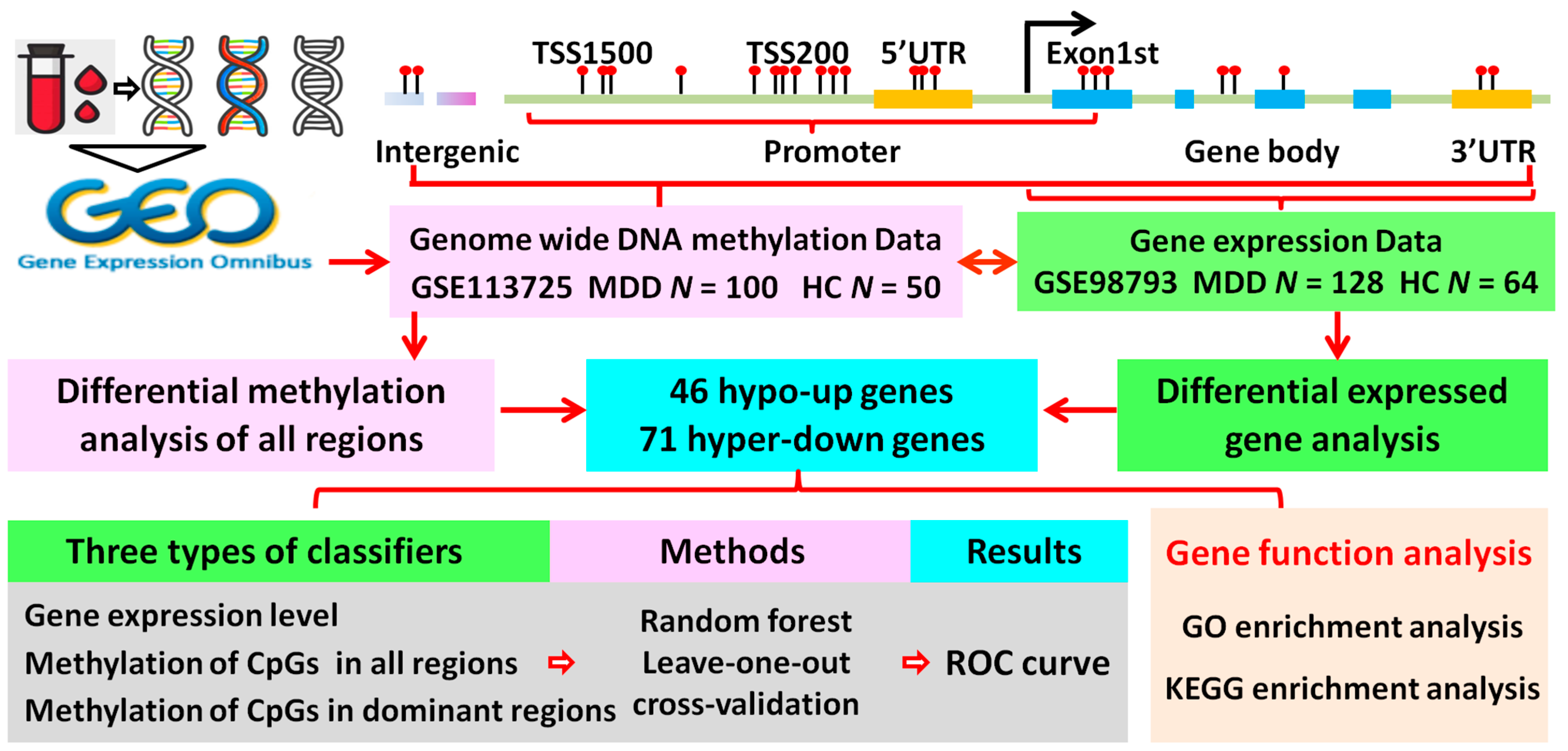
2. Materials and Methods
2.1. Data Collection
2.2. Screening of Differentially Expressed Genes (DEGs)
2.3. Differential Methylation Analysis
- (1) Methylation level of each CpG. The methylation level of each CpG can be calculated by the equation β = M / (M + U + a), where M > 0, U > 0, and a ≥ 0. M and U denote the number of methylated and unmethylated probes, respectively. Since M and U are small, “a” is set to 100 to stabilize the β-value [25].
- (2) Methylation level of different regions. In this study, we employed the “ChAMP” package (version 2.18.3) to measure the methylation level of the different regions (TSS1500, TSS200, 5′UTR, Exon1st, gene body, and 3′UTR) for each individual gene using the average β-value of the CpGs in the corresponding regions.
- (3) Methylation level of an individual gene. We also measured the methylation level of an individual gene using the average β-value of the CpGs in all regions.
- (4) Identification of differentially methylated genes (DMGs). To measure the methylation difference between MDD patients and healthy controls, a linear model was built. Ten quantiles of the delta beta value of all genes and all intergenic CpG sites were calculated, and the DMGs were defined as a ∆β value of <1/10 quantile or >9/10 quantile and a BH-adjusted p-value of <0.05.
2.4. Identification of the Dominant Hypo/Hyper-Methylated Regions
2.5. GO and KEGG Enrichment Analyses
2.6. Classifier Construction and LOO Validation
2.7. LOO Cross-Validation
3. Results
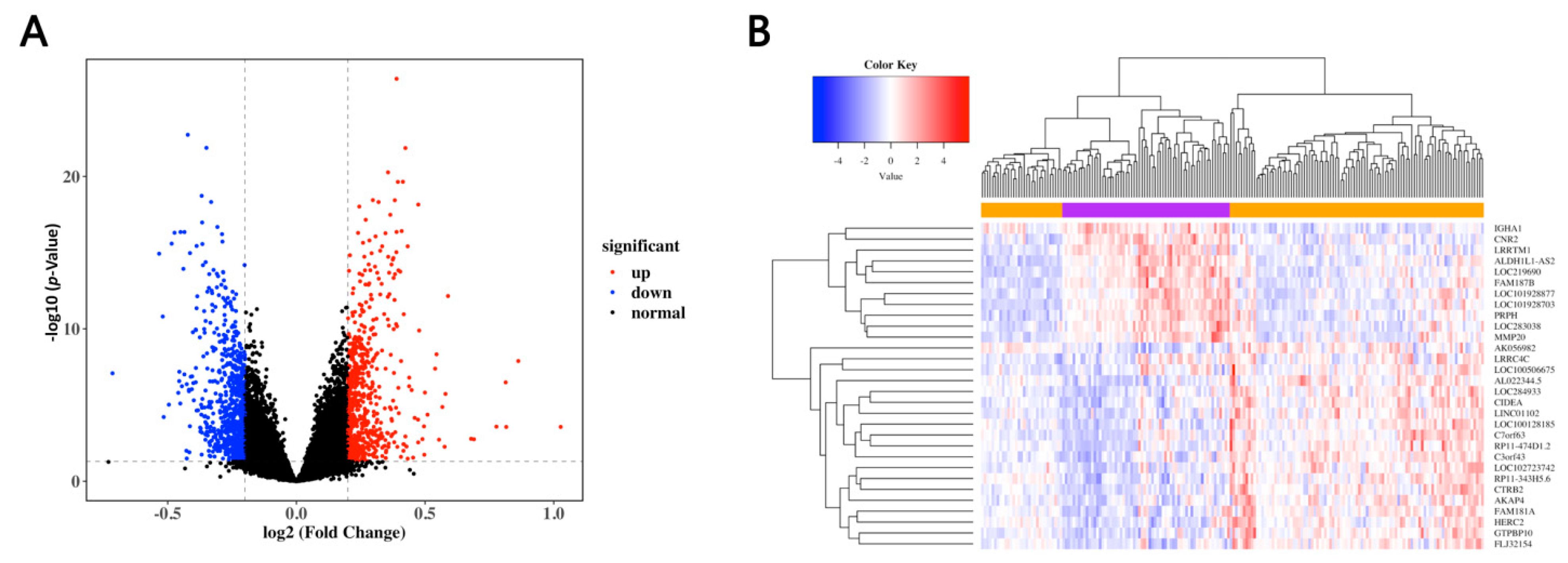
3.1. Identification of the DEGs in MDD
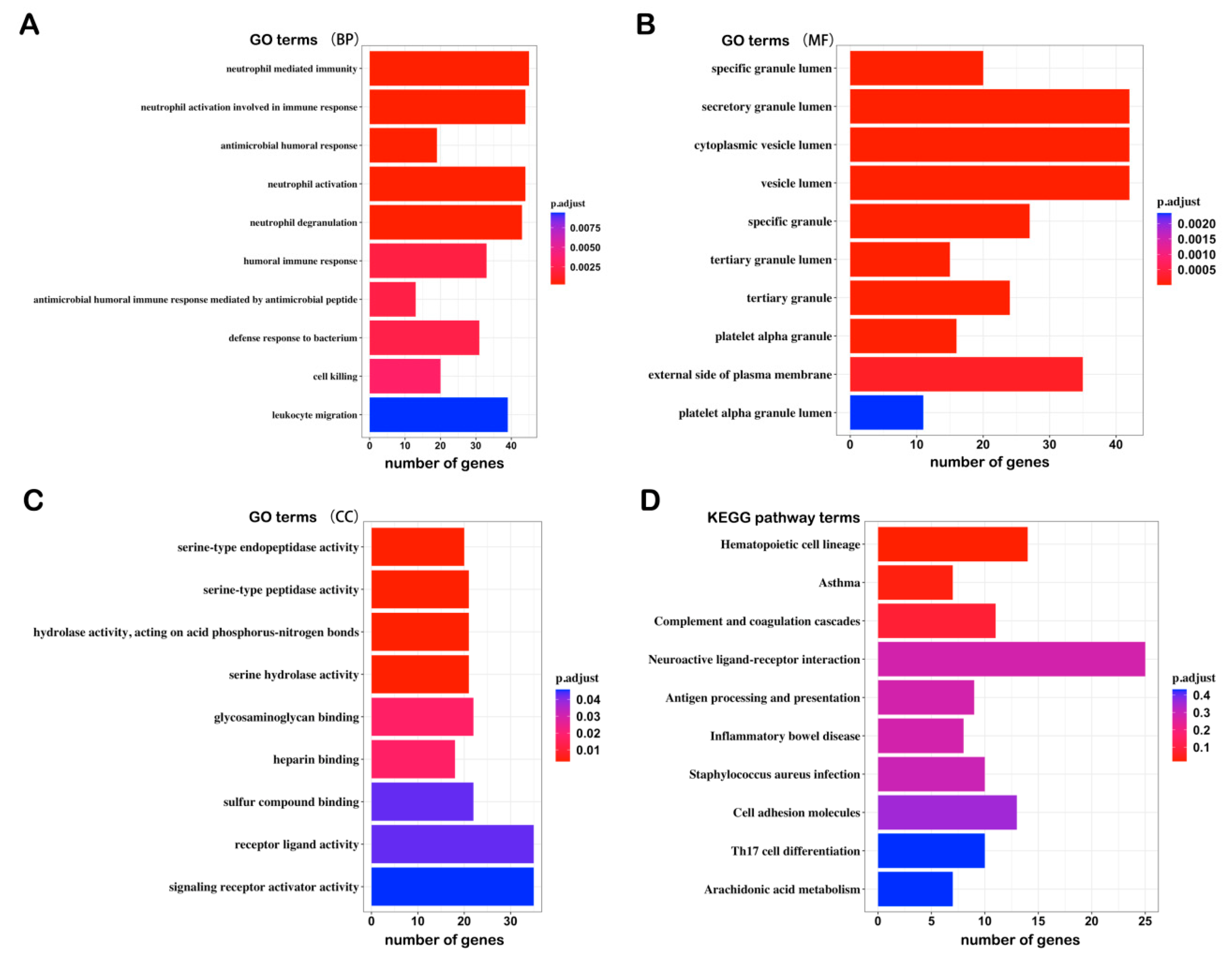
3.2. GO and KEGG Enrichment Analysis of the DEGs
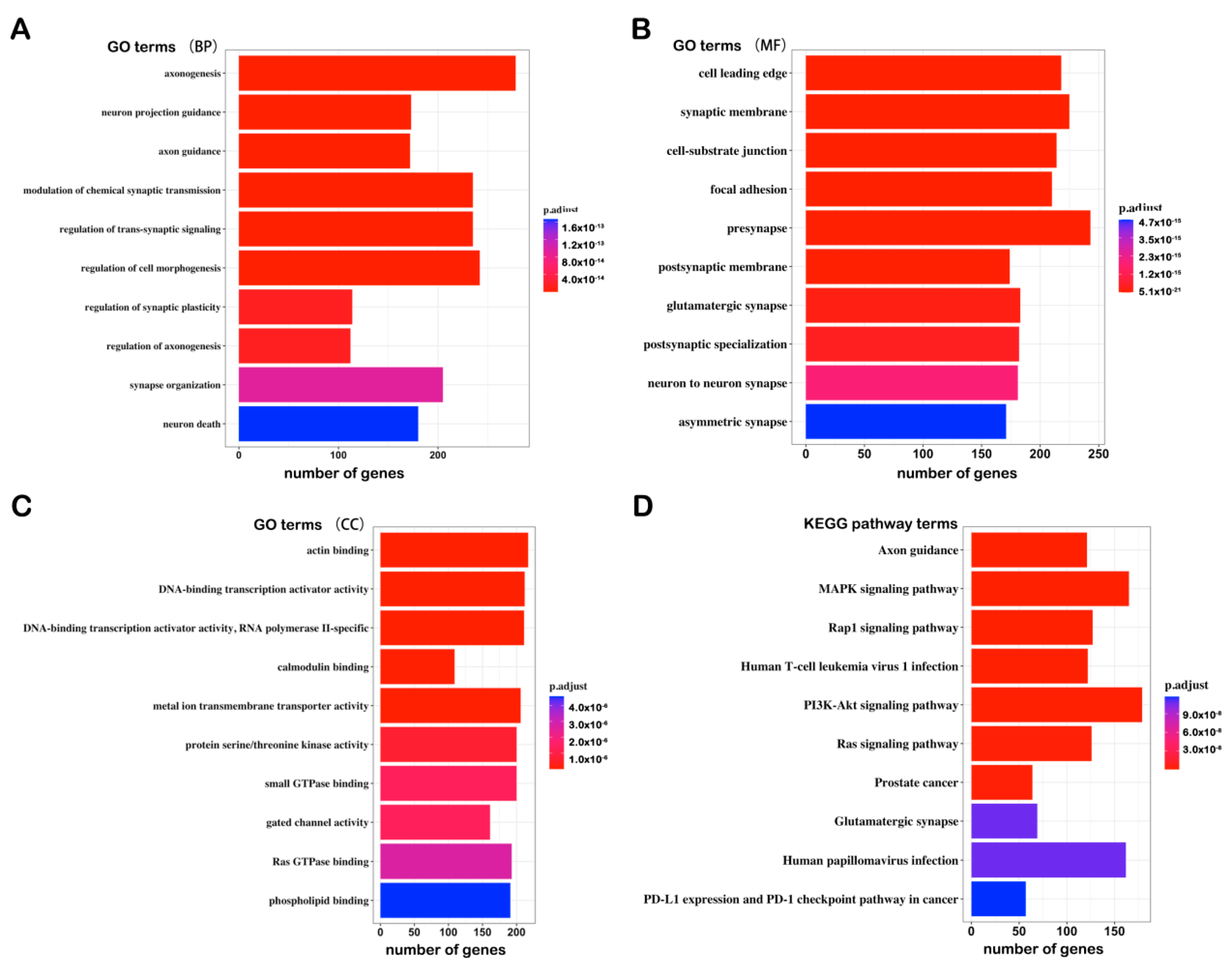
3.3. Identification and GO/KEGG Enrichment Analysis of the DMGs
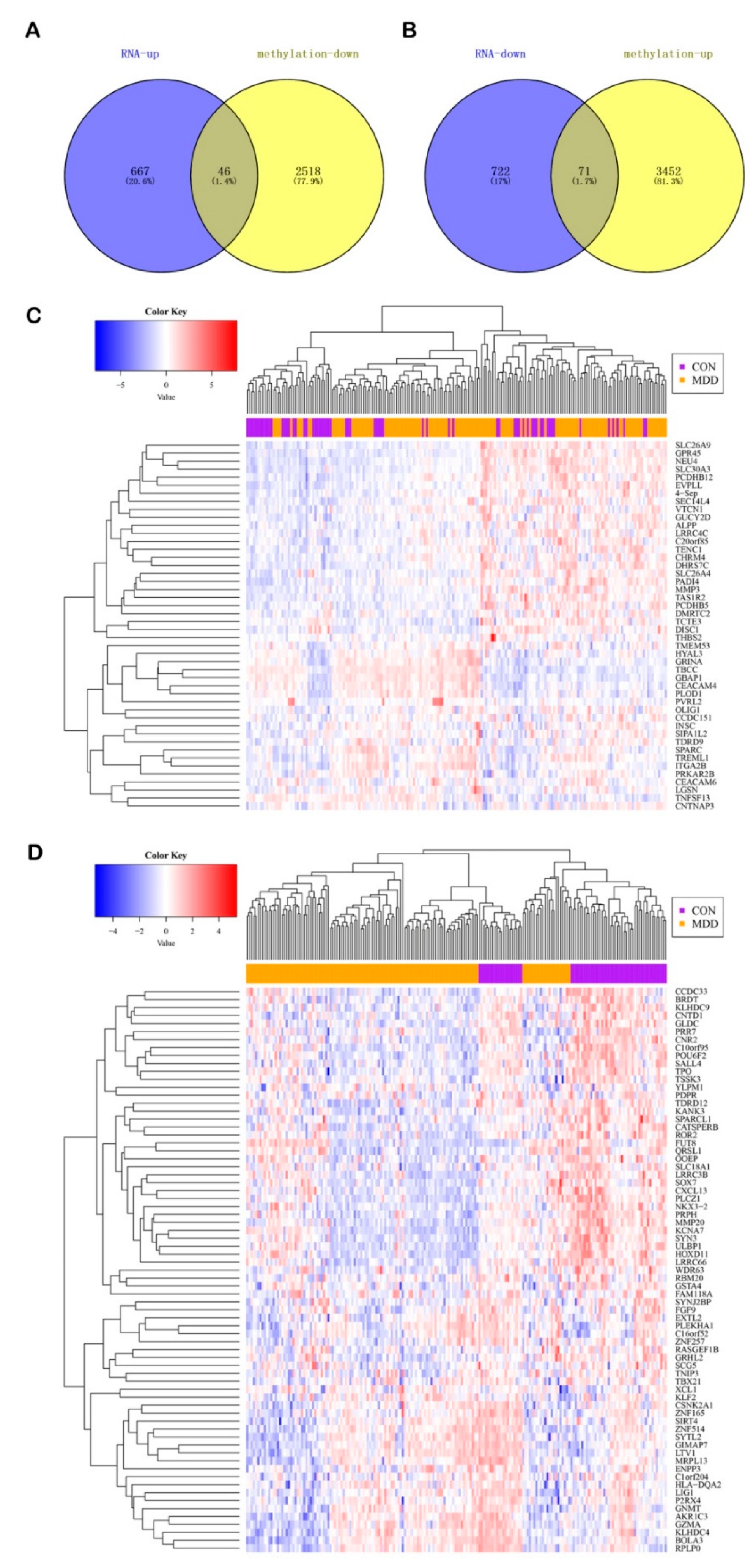
3.4. Integrated Analysis of the Gene Expression and DNA Methylation
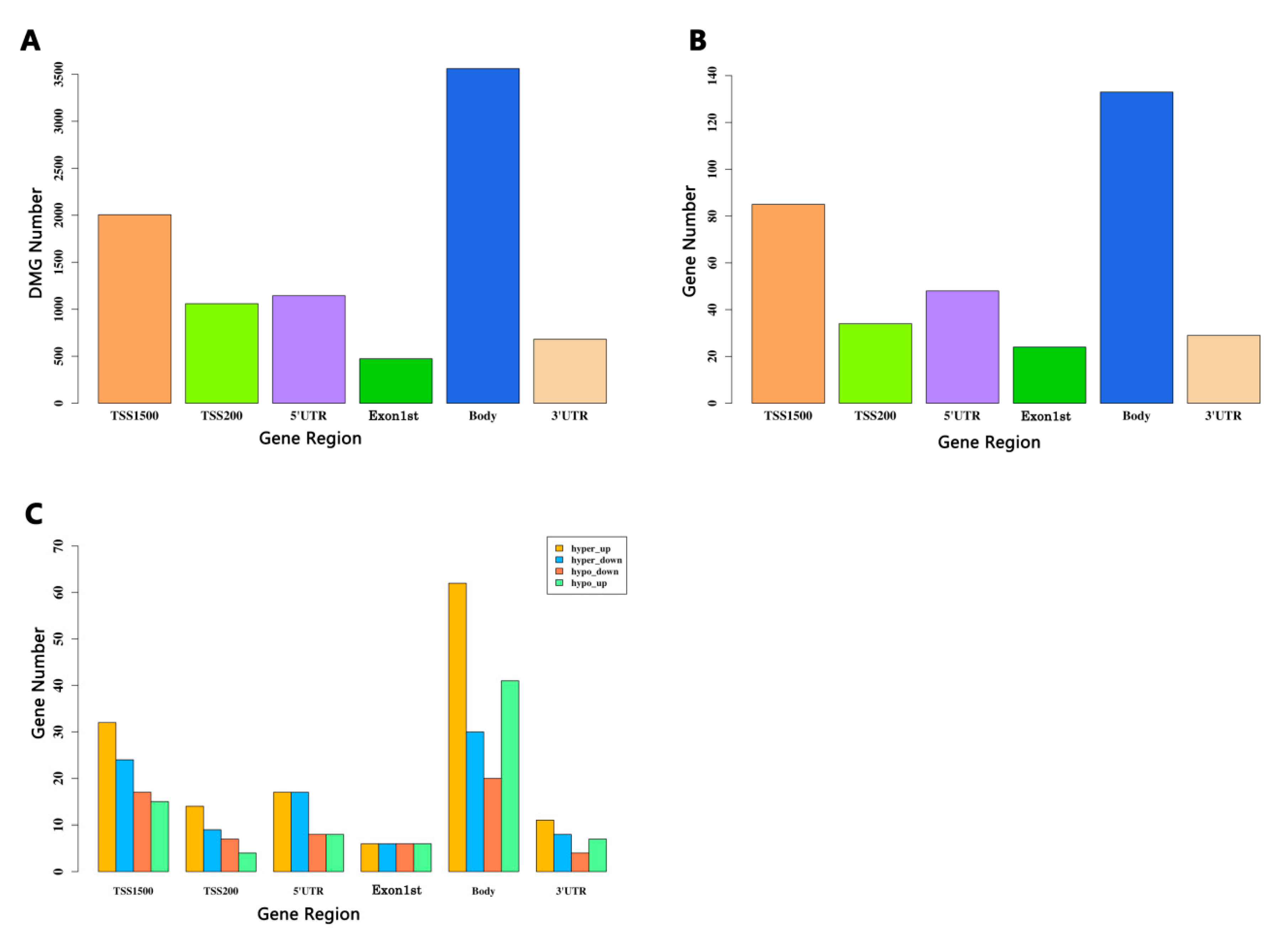
3.5. Identification of the DMGs Based on CpGs in the Different Regions
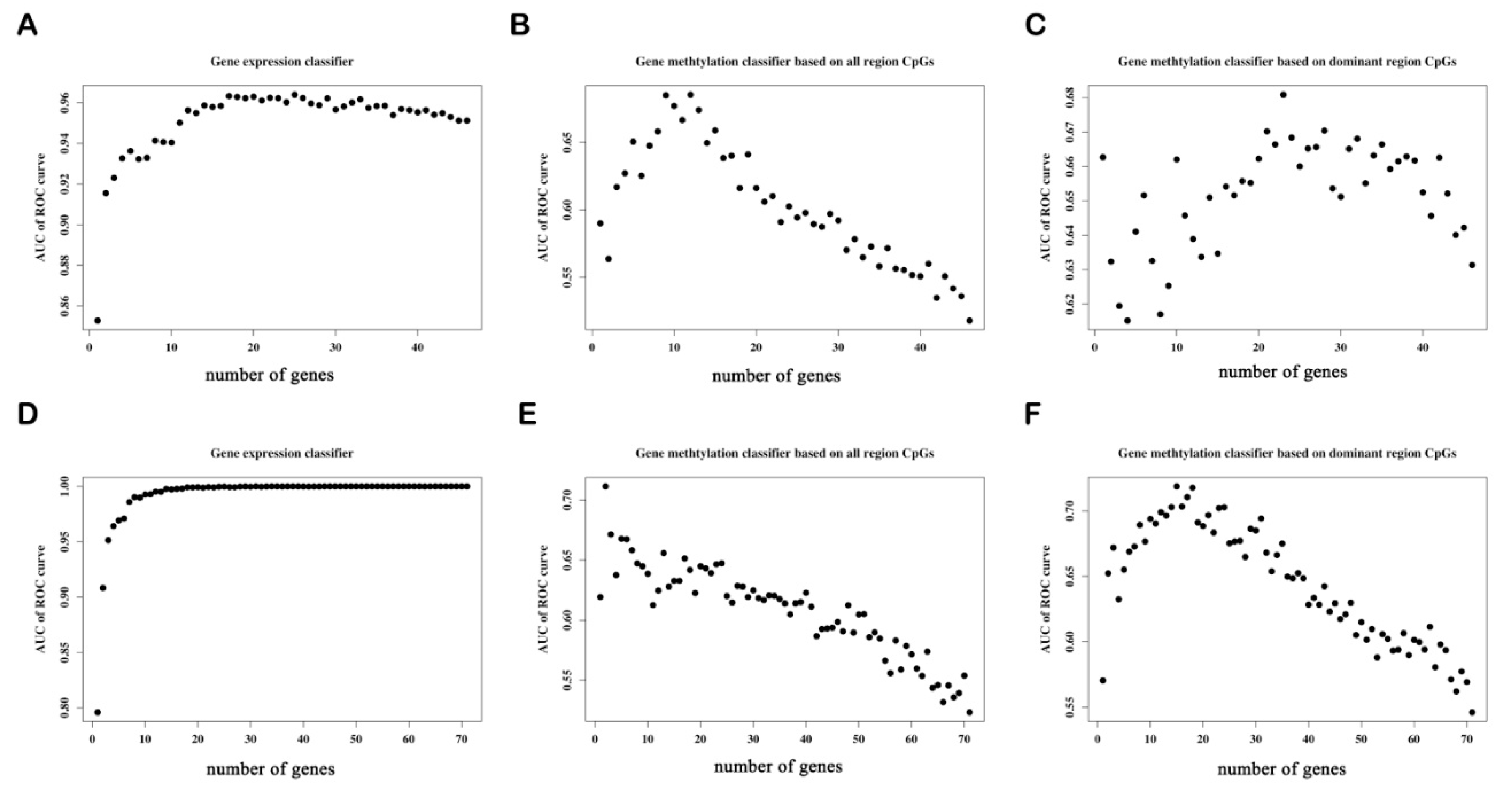
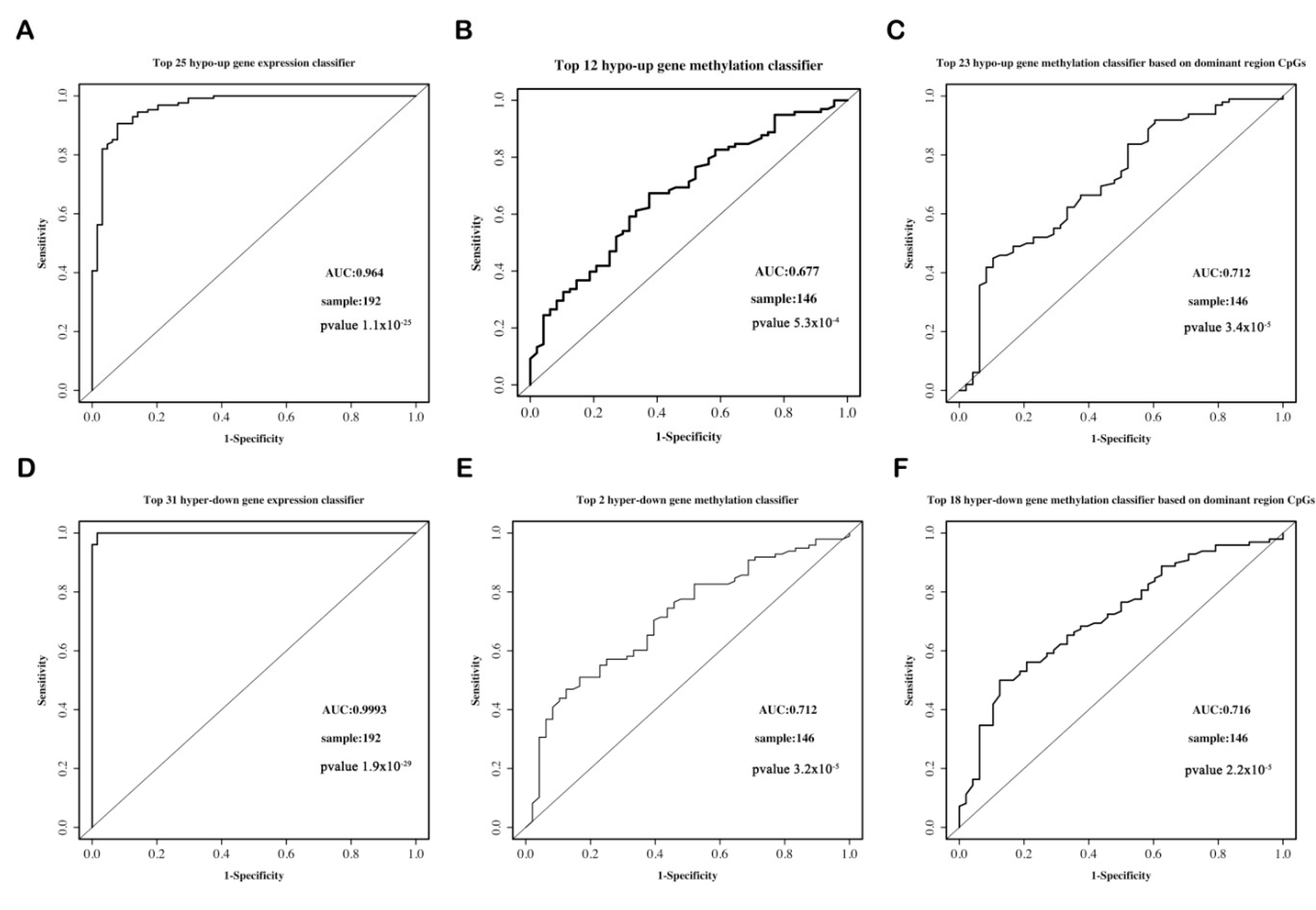
3.6. Classifier Construction and ROC Curve
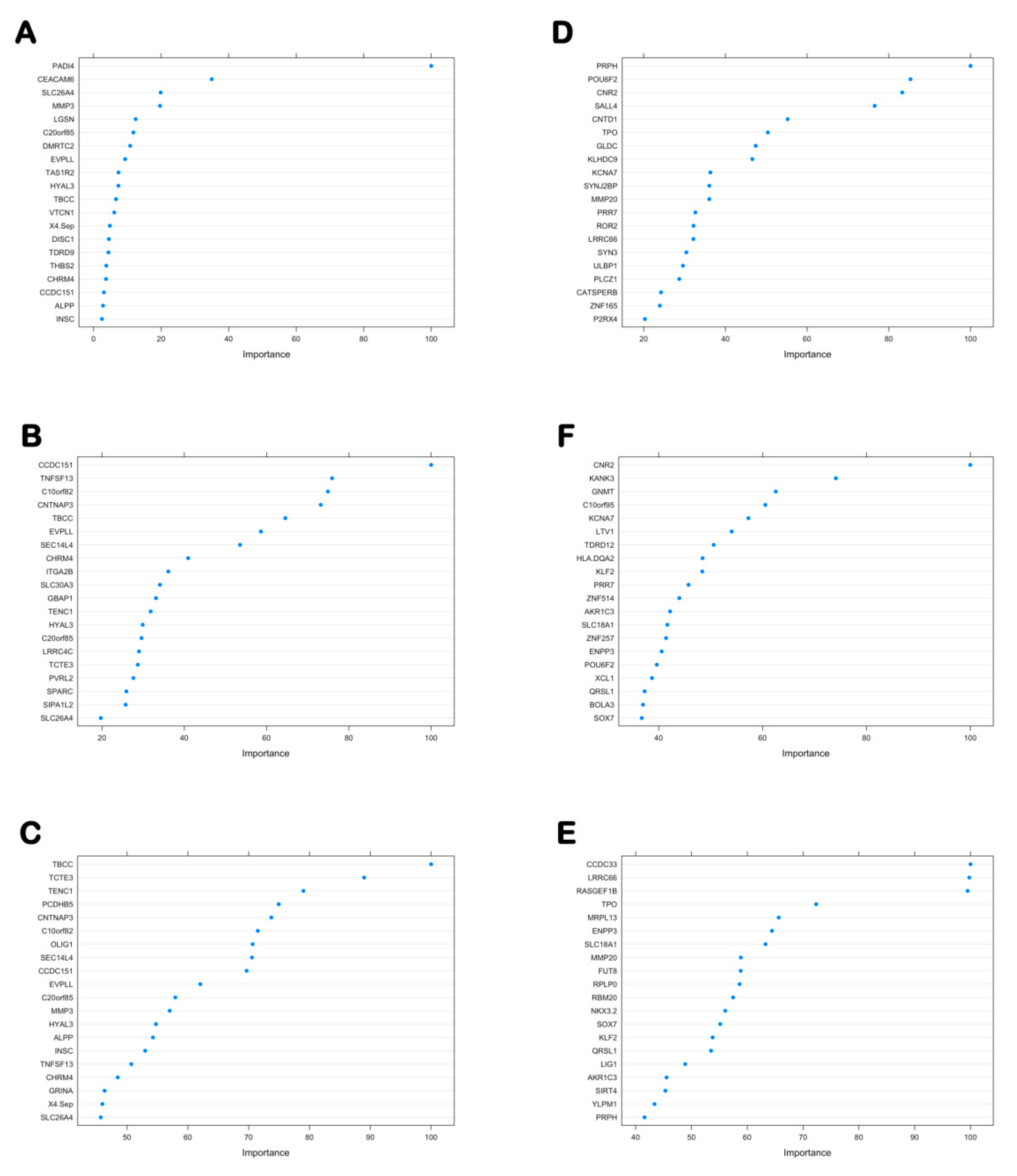
3.6.1. The Importance Score of the 46 Hypo-Up Genes and the 71 Hyper-Down Genes in Each Classifier
3.6.2. Determine the Number of Genes with the Best Predictive Power in Each Classifier
3.6.3. The Predictive Ability of Each Classifier
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Busch, Y.; Menke, A. Blood-based biomarkers predicting response to antidepressants. J. Neural Transm. 2019, 126, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Chirita, A.L.; Gheorman, V.; Bondari, D.; Rogoveanu, I. Current understanding of the neurobiology of major depressive disorder. Rom. J. Morphol. Embryol. 2015, 56, 651–658. [Google Scholar] [PubMed]
- Kennis, M.; Gerritsen, L.; van Dalen, M.; Williams, A.; Cuijpers, P.; Bockting, C. Prospective biomarkers of major depressive disorder: A systematic review and meta-analysis. Mol. Psychiatry 2019, 25, 321–338. [Google Scholar] [CrossRef]
- Hasin, D.S.; Sarvet, A.L.; Meyers, J.L.; Saha, T.D.; Ruan, W.J.; Stohl, M.; Grant, B.F. Epidemiology of adult DSM-5 major depressive disorder and its specifiers in the United States. JAMA Psychiatry 2018, 75, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Nemeroff, C.B. The burden of severe depression: A review of diagnostic challenges and treatment alternatives. J. Psychiatr. Res. 2007, 41, 189–206. [Google Scholar] [CrossRef]
- Siegle, G.J.; Carter, C.S.; Thase, M.E. Use of FMRI to predict recovery from unipolar depression with cognitive behavior therapy. Am. J. Psychiatry 2006, 163, 735–738. [Google Scholar] [CrossRef]
- Hepgul, N.; Cattaneo, A.; Zunszain, P.A.; Pariante, C.M. Depression pathogenesis and treatment: What can we learn from blood mRNA expression? BMC Med. 2013, 11, 28. [Google Scholar] [CrossRef]
- Tavakolizadeh, J.; Roshanaei, K.; Salmaninejad, A.; Yari, R.; Nahand, J.S.; Sarkarizi, H.K.; Mousavi, S.M.; Salarinia, R.; Rahmati, M.; Mousavi, S.F.; et al. MicroRNAs and exosomes in depression: Potential diagnostic biomarkers. J. Cell Biochem. 2018, 119, 3783–3797. [Google Scholar] [CrossRef]
- Chamberlain, S.R.; Cavanagh, J.; de Boer, P.; Mondelli, V.; Jones, D.N.C.; Drevets, W.C.; Cowen, P.J.; Harrison, N.A.; Pointon, L.; Pariante, C.M.; et al. Treatment-resistant depression and peripheral C-reactive protein. Br. J. Psychiatry 2019, 214, 11–19. [Google Scholar] [CrossRef]
- Lohoff, F.W. Overview of the genetics of major depressive disorder. Curr. Psychiatry Rep. 2010, 12, 539–546. [Google Scholar] [CrossRef]
- Sullivan, P.F.; Neale, M.C.; Kendler, K.S. Genetic epidemiology of major depression: Review and meta-analysis. Am. J. Psychiatry 2000, 157, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Kennedy, P.J.; Nestler, E.J. Epigenetics of the depressed brain: Role of histone acetylation and methylation. Neuropsychopharmacology 2013, 38, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; D’Arcy, C.; Li, X.; Zhang, T.; Joober, R.; Meng, X. What do DNA methylation studies tell us about depression? A systematic review. Transl. Psychiatry 2019, 9, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Klengel, T.; Pape, J.; Binder, E.B.; Mehta, D. The role of DNA methylation in stress-related psychiatric disorders. Neuropharmacology 2014, 80, 115–132. [Google Scholar] [CrossRef]
- Menke, A.; Binder, E.B. Epigenetic alterations in depression and antidepressant treatment. Dialogues Clin. Neurosci. 2014, 16, 395–404. [Google Scholar]
- Lisoway, A.J.; Zai, C.C.; Tiwari, A.K.; Kennedy, J.L. DNA methylation and clinical response to antidepressant medication in major depressive disorder: A review and recommendations. Neurosci. Lett. 2017, 669, 14–23. [Google Scholar] [CrossRef]
- Hüls, A.; Robins, C.; Conneely, K.N.; De Jager, P.L.; Bennett, D.A.; Epstein, M.P.; Wingo, T.S.; Wingo, A.P. Association between DNA methylation levels in brain tissue and late-life depression in community-based participants. Transl Psychiatry. 2020, 10, 262–271. [Google Scholar] [CrossRef]
- Clark, S.L.; Hattab, M.W.; Chan, R.F.; Shabalin, A.A.; Han, L.K.M.; Zhao, M.; Smit, J.H.; Jansen, R.; Milaneschi, Y.; Xie, L.Y.; et al. A methylation study of long-term depression risk. Mol. Psychiatry 2020, 25, 1334–1343. [Google Scholar] [CrossRef]
- Tseng, P.T.; Lin, P.Y.; Lee, Y.; Hung, C.F.; Lung, F.W.; Chen, C.S.; Chong, M. Age-associated decrease in global DNA methylation in patients with major depression. Neuropsychiatr. Dis. Treat. 2014, 10, 2105–2114. [Google Scholar]
- Uddin, M.; Koenen, K.C.; Aiello, A.E.; Wildman, D.E.; de los Santos, R.; Galea, S. Epigenetic and inflammatory marker profiles associated with depression in a community-based epidemiologic sample. Psychol. Med. 2011, 41, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Crawford, B.; Craig, Z.; Mansell, G.; White, I.; Smith, A.; Spaull, S.; Imm, J.; Hannon, E.; Wood, A.; Yaghootkar, H.; et al. DNA methylation and inflammation marker profiles associated with a history of depression. Hum. Mol. Genet. 2018, 27, 2840–2850. [Google Scholar] [CrossRef] [PubMed]
- Leday, G.G.R.; Vertes, P.E.; Richardson, S.; Greene, J.R.; Regan, T.; Khan, S.; Henderson, R.; Freeman, T.C.; Pariante, C.M.; Harrison, N.A.; et al. Replicable and coupled changes in innate and adaptive immune gene expression in two case-control studies of blood microarrays in major depressive disorder. Biol. Psychiatry 2018, 83, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Weinhold, L.; Wahl, S.; Pechlivanis, S.; Hoffmann, P.; Schmid, M. A statistical model for the analysis of Beta values in DNA methylation studies. BMC Bioinform. 2016, 17, 480–490. [Google Scholar] [CrossRef]
- Wang, C.; Chen, L.; Yang, Y.; Zhang, M.; Wong, G. Identification of potential blood biomarkers for Parkinson’s disease by gene expression and DNA methylation data integration analysis. Clin. Epigenet. 2019, 11, 24–39. [Google Scholar] [CrossRef]
- Xu, T.; Li, B.; Zhao, M.; Szulwach, K.E.; Street, R.C.; Lin, L.; Yao, B.; Zhang, F.; Jin, P.; Wu, H.; et al. Base-resolution methylation patterns accurately predict transcription factor bindings in vivo. Nucleic Acids Res. 2015, 43, 2757–2766. [Google Scholar] [CrossRef]
- Rajagopal, N.; Xie, W.; Li, Y.; Wagner, U.; Wang, W.; Stamatoyannopoulos, J.; Ernst, J.; Kellis, M.; Ren, B. RFECS: A random-forest based algorithm for enhancer identification from chromatin state. PLoS Comput. Biol. 2013, 9, e1002968. [Google Scholar] [CrossRef]
- Bufalino, C.; Hepgul, N.; Aguglia, E.; Pariante, C.M. The role of immune genes in the association between depression and inflammation: A review of recent clinical studies. Brain Behav. Immun. 2013, 31, 31–47. [Google Scholar] [CrossRef]
- Debnath, M.; Doyle, K.M.; Langan, C.; McDonald, C.; Leonard, B.; Cannon, D.M. Recent advances in psychoneuroimmunology: Inflammation in psychiatric disorders. Transl. Neurosci. 2011, 2, 121–137. [Google Scholar] [CrossRef]
- Messay, B.; Lim, A.; Marsland, A.L. Current understanding of the bi-directional relationship of major depression with inflammation. Biol. Mood Anxiety Disor. 2012, 2, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Hiles, S.A.; Bake, R.A.L.; de Malmanche, T.; Attia, J. A meta-analysis of differences in IL-6 and IL-10 between people with and without depression: Exploring the causes of heterogeneity. Brain Behav. Immun. 2012, 2626, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Freud, S. Mourning and Melancholia. In The Standard Edition of the Complete Psychological Works of Sigmund Freud; Freud, S., Strachey, J., Eds.; Hogarth Press: London, UK, 1957; Volume 14, pp. 243–258. [Google Scholar]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Slavich, G.M.; Irwin, M.R. From stress to inflammation and major depressive disorder: A social signal transduction theory of depression. Psychol. Bull. 2014, 140, 774–815. [Google Scholar] [CrossRef]
- Kempton, M.J.; Salvador, Z.; Munafo, M.R.; Geddes, J.R.; Simmons, A.; Frangou, S.; Williams, S.C. Structural neuroimaging studies in major depressive disorder. Meta-analysis and comparison with bipolar disorder. Arch. Gen. Psychiatry 2011, 68, 675–690. [Google Scholar] [CrossRef]
- Cobb, J.A.; Simpson, J.; Mahajan, G.J.; Overholser, J.C.; Jurjus, G.; Dieter, L.; Herbst, N.; May, W.; Rajkowska, G.; Stockmeier, C.A. Hippocampal volume and total cell numbers in major depressive disorder. J. Psychiatr. Res. 2013, 47, 299–306. [Google Scholar] [CrossRef]
- Alfonso, J.; Frick, L.R.; Silberman, D.M.; Palumbo, M.L.; Gerano, A.M.; Frasch, A.C. Regulation of hippocampal gene expression is conserved in two species subjected to different stressors and antidepressant treatment. Biol. Psychiatry 2006, 59, 244–251. [Google Scholar] [CrossRef]
- Patel, M.N.; McNamara, J.O. Selective enhancement of axonal branching of cultured dentate gyrus neurons by neurotrophic factors. Neuroscience 1995, 69, 763–770. [Google Scholar] [CrossRef]
- Januar, V.; Ancelin, M.L.; Ritchie, K.; Saffery, R.; Ryan, J. BDNF promoter methylation and genetic variation in late-life depression. Transl. Psychiatry 2015, 5, e619. [Google Scholar] [CrossRef]
- Wang, J.; Hodes, G.E.; Zhang, H.; Zhang, S.; Zhao, W.; Golden, S.A.; Bi, W.; Menard, C.; Kana, V.; Leboeuf, M.; et al. Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nat. Commun. 2018, 9, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Sosa, M.A.G.; Huang, L.H.; Midgett, R.M.; Kuo, K.C.; McCune, R.A.; Gehrk, E.C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982, 10, 2709–2721. [Google Scholar] [CrossRef]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Toperoff, G.; Rosenberg, M.; Hellman, A. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 2011, 20, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Tozzi, L.; Farrell, C.; Booij, L.; Doolin, K.; Nemoda, Z.; Szyf, M.; Pomares, F.B.; Chiarella, J.; O’Keane, V.; Frodl, T. Epigenetic changes of FKBP5 as a link connecting genetic and environmental risk factors with structural and functional brain changes in major depression. Neuropsychopharmacology 2018, 43, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, K.L.; Moore, S.R.; Davis, E.G.; MacIsaac, J.L.; Lin, D.T.S.; Kobor, M.S.; Gotlib, I.H. DNA methylation of HPA-axis genes and the onset of major depressive disorder in adolescent girls: A prospective analysis. Transl. Psychiatry 2019, 9, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Sukjai, B.T.; Suttajit, S.; Thanoi, S.; Dalton, C.F.; Reynolds, G.P.; Nudmamud-Thanoi, S. Parvalbumin promoter methylation altered in major depressive disorder. Int. J. Med. Sci. 2019, 16, 1207–1214. [Google Scholar] [CrossRef]
- Lam, D.; Ancelin, M.L.; Ritchie, K.; Poli, R.F.; Saffery, R.; Ryan, J. Genotype-dependent associations between serotonin transporter gene (SLC6A4) DNA methylation and late-life depression. BMC Psychiatry 2018, 18, 282. [Google Scholar] [CrossRef]
- Peng, Y.; Wu, Q.; Wang, L.; Wang, H.; Yin, F. A DNA methylation signature to improve survival prediction of gastric cancer. Clin. Epigenetics. 2020, 12, 15–30. [Google Scholar] [CrossRef]
- Griffith, O.L.; Pepin, F.; Enache, O.M.; Heiser, L.M.; Collisson, E.A.; Spellman, P.T.; Gray, J.W. A robust prognostic signature for hormone-positive node-negative breast cancer. Genome Med. 2013, 5, 92–105. [Google Scholar] [CrossRef]
- Zeng, D.; Li, M.; Zhou, R.; Zhang, J.; Sun, H.; Shi, M.; Bin, J.; Liao, Y.; Rao, J.; Liao, W. Tumor Microenvironment Characterization in Gastric Cancer Identifies Prognostic and Immunotherapeutically Relevant Gene Signatures. Cancer Immunol. Res. 2019, 7, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Lee, J.; Akter, S.; Rogers, M.; Grene, R.; Li, S. Prediction of condition-specific regulatory genes using machine learning. Nucleic Acids Res. 2020, 48, e62. [Google Scholar] [CrossRef] [PubMed]
- Shaik, R.; Ramakrishna, W. Machine learning approaches distinguish multiple stress conditions using stress-responsive genes and identify candidate genes for broad resistance in rice. Plant. Physiol. 2014, 164, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.N.; Lee, T.Y.; Hung, Y.C.; Li, G.Z.; Tseng, K.C.; Liu, Y.H.; Kuo, P.L.; Zheng, H.Q.; Chang, W.C. PlantPAN3.0: A new and updated resource for reconstructing transcriptional regulatory networks from ChIP-seq experiments in plants. Nucleic Acids Res. 2019, 47, D1155–D1163. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Direction | Gene Symbol |
|---|---|
| hypo-up | SEPT4, ALPP, C20orf85, CCDC151, CEACAM4, CEACAM6, CHRM4, CNTNAP3, DHRS7C, DISC1, DMRTC2, EVPLL, GBAP1, GPR45, GRINA, GUCY2D, HYAL3, INSCI, TGA2B, LGSN, LRRC4C, MMP3, NEU4, OLIG1, PADI4, PCDHB12, PCDHB5, PLOD1, PRKAR2B, PVRL2, SEC14L4, SIPA1L2, SLC26A4, SLC26A9, SLC30A3, SPARC, TAS1R2, TBCC, TCTE3, TDRD9, TENC1, THBS2, TMEM53, TNFSF13, TREML1, VTCN1 |
| hyper-down | AKR1C3, BOLA3, BRDT, C10orf95, C16orf52, C1orf204, CATSPERB, CCDC33, CNR2, CNTD1, CSNK2A1CXCL13, ENPP3, EXTL2, FAM118A, FGF9 FUT8, GIMAP7, GLDC, GNMT, GRHL2, GSTA4, GZMA HLA-DQA2, HOXD11, KANK3, KCNA7, KLF2 KLHDC4, KLHDC9, LIG1, LRRC3B, LRRC66, LTV1, MMP20, MRPL13, NKX3-2, OOEP, P2RX4, PDPR, PLCZ1, PLEKHA1, POU6F2, PRPH, PRR7, QRSL1, RASGEF1B, RBM20, ROR2, RPLP0, SALL4, SCG5, SIRT4, SLC18A1, SOX7, SPARCL1, SYN3, SYNJ2BP, SYTL2, TBX21, TDRD12, TNIP3, TPO, TSSK3, ULBP1, WDR63, XCL1, YLPM1, ZNF165ZNF257, ZNF514 |
| Group | TSS1500 | TSS200 | 5′UTR | Exon1st | Body | 3′UTR |
|---|---|---|---|---|---|---|
| hypo-up | 15 | 4 | 8 | 6 | 41 | 7 |
| hypo-down | 17 | 7 | 8 | 6 | 20 | 4 |
| hyper-up | 32 | 14 | 17 | 6 | 62 | 11 |
| hyper-down | 24 | 9 | 17 | 6 | 30 | 8 |
| Importance of 46 Hypo-Up Genes Based on Gene Expression Level | Importance of 46 Hypo-Up Genes Based on DNA Methylation Level of All Regions | Importance of 46 Hypo-Up Genes Based on DNA Methylation Level of Dominant Regions | |||
|---|---|---|---|---|---|
| Gene Symbol | Importance | Gene Symbol | Importance | Gene Symbol | Importance |
| PADI4 | 100.00 | CCDC151 | 100.00 | TBCC | 100.00 |
| CEACAM6 | 34.98 | TNFSF13 | 75.91 | TCTE3 | 88.98 |
| SLC26A4 | 19.87 | C10orf82 | 74.91 | TENC1 | 79.01 |
| MMP3 | 19.66 | CNTNAP3 | 73.18 | PCDHB5 | 74.94 |
| LGSN | 12.47 | TBCC | 64.55 | CNTNAP3 | 73.72 |
| C20orf85 | 11.79 | EVPLL | 58.62 | C10orf82 | 71.51 |
| DMRTC2 | 10.84 | SEC14L4 | 53.55 | OLIG1 | 70.65 |
| EVPLL | 9.34 | CHRM4 | 40.94 | SEC14L4 | 70.53 |
| TAS1R2 | 7.39 | ITGA2B | 36.11 | CCDC151 | 69.67 |
| HYAL3 | 7.34 | SLC30A3 | 34.08 | EVPLL | 62.05 |
| TBCC | 6.63 | GBAP1 | 33.11 | C20orf85 | 57.94 |
| VTCN1 | 6.12 | TENC1 | 31.83 | MMP3 | 57.02 |
| X4.SEP | 4.80 | HYAL3 | 29.89 | HYAL3 | 54.74 |
| DISC1 | 4.52 | C20orf85 | 29.60 | ALPP | 54.27 |
| TDRD9 | 4.39 | LRRC4C | 28.99 | INSC | 52.99 |
| THBS2 | 3.75 | TCTE3 | 28.70 | TNFSF13 | 50.71 |
| CHRM4 | 3.68 | PVRL2 | 27.63 | CHRM4 | 48.47 |
| CCDC151 | 3.04 | SPARC | 25.92 | GRINA | 46.32 |
| ALPP | 2.79 | SIPA1L2 | 25.75 | X4.SEP | 45.95 |
| INSC | 2.47 | SLC26A4 | 19.70 | SLC26A4 | 45.69 |
| GPR45 | 2.12 | OLIG1 | 19.45 | PVRL2 | 44.90 |
| PCDHB5 | 2.09 | GPR45 | 19.07 | TDRD9 | 39.39 |
| ITGA2B | 2.01 | PCDHB12 | 18.70 | DHRS7C | 38.25 |
| PLOD1 | 2.00 | X4.Sep | 17.39 | PCDHB12 | 38.25 |
| SLC30A3 | 1.92 | TAS1R2 | 17.30 | PADI4 | 35.12 |
| PRKAR2B | 1.84 | SLC26A9 | 17.11 | GBAP1 | 34.98 |
| TMEM53 | 1.69 | CEACAM4 | 16.23 | CEACAM6 | 33.94 |
| PCDHB12 | 1.58 | CEACAM6 | 16.17 | LGSN | 32.33 |
| GUCY2D | 1.52 | VTCN1 | 16.08 | GUCY2D | 31.03 |
| TCTE3 | 1.32 | MMP3 | 15.67 | PLOD1 | 30.28 |
| CEACAM4 | 1.27 | TDRD9 | 15.51 | PRKAR2B | 29.78 |
| TNFSF13 | 1.26 | NEU4 | 15.48 | NEU4 | 29.59 |
| CNTNAP3 | 1.26 | DMRTC2 | 14.53 | THBS2 | 28.19 |
| LRRC4C | 1.13 | PADI4 | 13.87 | SIPA1L2 | 26.04 |
| SEC14L4 | 1.07 | GUCY2D | 12.33 | GPR45 | 25.63 |
| DHRS7C | 1.07 | ALPP | 10.95 | VTCN1 | 20.80 |
| SLC26A9 | 1.04 | PLOD1 | 10.59 | TMEM53 | 18.90 |
| OLIG1 | 0.98 | PCDHB5 | 9.36 | DMRTC2 | 18.00 |
| GRINA | 0.96 | THBS2 | 9.34 | LRRC4C | 11.03 |
| PVRL2 | 0.83 | PRKAR2B | 8.81 | SLC26A9 | 10.08 |
| TENC1 | 0.74 | DHRS7C | 7.63 | ITGA2B | 8.65 |
| NEU4 | 0.59 | GRINA | 7.02 | SPARC | 8.55 |
| SIPA1L2 | 0.53 | TMEM53 | 6.53 | CEACAM4 | 8.35 |
| SPARC | 0.43 | DISC1 | 3.23 | SLC30A3 | 7.98 |
| TREML1 | 0.28 | LGSN | 2.52 | TAS1R2 | 5.77 |
| GBAP1 | 0.00 | INSC | 0.00 | DISC1 | 0.00 |
| Importance of 71 Hyper-Down Genes Based on Gene Expression Level | Importance of 71 Hyper-Down Genes Based on DNA Methylation Level of All Regions | Importance of 71 Hyper-Down Genes Based on DNA Methylation Level of Dominant Regions | |||
|---|---|---|---|---|---|
| Gene Symbol | Importance | Gene Symbol | Importance | Gene Symbol | Importance |
| PRPH | 100.00 | CNR2 | 100.00 | CCDC33 | 100.00 |
| POU6F2 | 85.32 | KANK3 | 74.12 | LRRC66 | 99.79 |
| CNR2 | 83.29 | GNMT | 62.55 | RASGEF1B | 99.50 |
| SALL4 | 76.54 | C10orf95 | 60.54 | TPO | 72.35 |
| CNTD1 | 55.23 | KCNA7 | 57.28 | MRPL13 | 65.64 |
| TPO | 50.37 | LTV1 | 54.06 | ENPP3 | 64.41 |
| GLDC | 47.41 | TDRD12 | 50.59 | SLC18A1 | 63.26 |
| KLHDC9 | 46.57 | HLA.DQA2 | 48.45 | MMP20 | 58.84 |
| KCNA7 | 36.31 | KLF2 | 48.39 | FUT8 | 58.82 |
| SYNJ2BP | 36.05 | PRR7 | 45.75 | RPLP0 | 58.62 |
| MMP20 | 36.04 | ZNF514 | 43.97 | RBM20 | 57.46 |
| PRR7 | 32.64 | AKR1C3 | 42.20 | NKX3.2 | 56.05 |
| ROR2 | 32.18 | SLC18A1 | 41.67 | SOX7 | 55.14 |
| LRRC66 | 32.14 | ZNF257 | 41.42 | KLF2 | 53.79 |
| SYN3 | 30.43 | ENPP3 | 40.57 | QRSL1 | 53.50 |
| ULBP1 | 29.59 | POU6F2 | 39.64 | LIG1 | 48.87 |
| PLCZ1 | 28.68 | XCL1 | 38.70 | AKR1C3 | 45.54 |
| CATSPERB | 24.22 | QRSL1 | 37.29 | SIRT4 | 45.32 |
| ZNF165 | 23.93 | BOLA3 | 36.98 | YLPM1 | 43.38 |
| P2RX4 | 20.28 | SOX7 | 36.74 | PRPH | 41.58 |
| GIMAP7 | 19.69 | FGF9 | 34.30 | PLEKHA1 | 41.24 |
| NKX3.2 | 19.57 | SYTL2 | 34.13 | SYNJ2BP | 40.67 |
| CSNK2A1 | 19.37 | TBX21 | 33.91 | FGF9 | 40.42 |
| LIG1 | 18.73 | MRPL13 | 33.58 | KLHDC9 | 39.15 |
| C10orf95 | 18.64 | TNIP3 | 32.47 | SYTL2 | 37.12 |
| HOXD11 | 14.99 | C1orf204 | 32.40 | CATSPERB | 35.66 |
| MRPL13 | 14.29 | EXTL2 | 32.37 | GSTA4 | 31.31 |
| BRDT | 12.75 | GRHL2 | 30.95 | GNMT | 30.86 |
| SYTL2 | 12.32 | MMP20 | 30.93 | KANK3 | 30.20 |
| RBM20 | 12.02 | RASGEF1B | 30.61 | ROR2 | 28.88 |
| SPARCL1 | 11.97 | CATSPERB | 29.19 | P2RX4 | 28.65 |
| ZNF514 | 11.36 | C16orf52 | 27.04 | CXCL13 | 27.97 |
| GRHL2 | 11.27 | SIRT4 | 25.34 | HLA.DQA2 | 27.92 |
| SLC18A1 | 11.15 | GLDC | 24.13 | BOLA3 | 26.26 |
| GNMT | 10.75 | CXCL13 | 24.12 | LTV1 | 25.90 |
| LRRC3B | 10.62 | FUT8 | 22.93 | GIMAP7 | 25.73 |
| GZMA | 10.06 | SYNJ2BP | 21.97 | FAM118A | 25.50 |
| FAM118A | 9.29 | KLHDC9 | 21.81 | GRHL2 | 24.98 |
| TDRD12 | 9.01 | KLHDC4 | 21.53 | CSNK2A1 | 24.97 |
| RPLP0 | 8.89 | GIMAP7 | 20.03 | HOXD11 | 24.96 |
| CXCL13 | 8.53 | TSSK3 | 19.54 | BRDT | 24.18 |
| CCDC33 | 8.53 | HOXD11 | 18.13 | C10orf95 | 23.17 |
| ZNF257 | 8.03 | BRDT | 17.55 | C1orf204 | 23.08 |
| C1orf204 | 7.90 | PRPH | 16.54 | XCL1 | 23.02 |
| BOLA3 | 7.60 | CSNK2A1 | 15.97 | TSSK3 | 22.30 |
| TSSK3 | 7.59 | TPO | 15.85 | CNR2 | 21.92 |
| KANK3 | 7.51 | NKX3.2 | 14.99 | TNIP3 | 21.08 |
| ENPP3 | 6.89 | RBM20 | 14.11 | WDR63 | 20.39 |
| GSTA4 | 6.80 | PDPR | 13.91 | OOEP | 19.69 |
| AKR1C3 | 6.75 | LRRC66 | 12.73 | KLHDC4 | 19.50 |
| KLHDC4 | 6.28 | SPARCL1 | 12.28 | GLDC | 19.26 |
| EXTL2 | 5.85 | CNTD1 | 12.28 | SPARCL1 | 19.17 |
| SIRT4 | 5.50 | FAM118A | 12.22 | GZMA | 18.74 |
| FGF9 | 5.22 | CCDC33 | 11.64 | PLCZ1 | 18.74 |
| SCG5 | 4.88 | YLPM1 | 11.39 | SCG5 | 17.37 |
| FUT8 | 4.51 | RPLP0 | 9.51 | POU6F2 | 16.12 |
| OOEP | 4.45 | PLEKHA1 | 9.49 | PDPR | 15.66 |
| TNIP3 | 4.44 | LIG1 | 9.40 | PRR7 | 15.27 |
| PDPR | 4.43 | LRRC3B | 9.20 | EXTL2 | 14.63 |
| PLEKHA1 | 4.29 | ZNF165 | 8.61 | ZNF165 | 13.83 |
| SOX7 | 3.97 | GSTA4 | 7.98 | LRRC3B | 13.70 |
| C16orf52 | 3.90 | SYN3 | 7.58 | SYN3 | 13.64 |
| RASGEF1B | 3.56 | SALL4 | 7.46 | SALL4 | 13.33 |
| WDR63 | 3.54 | SCG5 | 6.31 | ULBP1 | 12.34 |
| TBX21 | 2.92 | ULBP1 | 6.12 | TDRD12 | 12.16 |
| HLA.DQA2 | 2.77 | OOEP | 5.27 | ZNF514 | 8.22 |
| KLF2 | 2.76 | WDR63 | 4.40 | C16orf52 | 7.70 |
| LTV1 | 2.35 | PLCZ1 | 3.34 | TBX21 | 7.52 |
| QRSL1 | 1.46 | GZMA | 2.84 | CNTD1 | 7.11 |
| XCL1 | 0.72 | ROR2 | 0.52 | KCNA7 | 5.55 |
| YLPM1 | 0.00 | P2RX4 | 0.00 | ZNF257 | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Y.; Xiao, L.; Chen, L.; Zheng, Y.; Zhang, C.; Wang, G. Integrated Analysis of Methylomic and Transcriptomic Data to Identify Potential Diagnostic Biomarkers for Major Depressive Disorder. Genes 2021, 12, 178. https://doi.org/10.3390/genes12020178
Xie Y, Xiao L, Chen L, Zheng Y, Zhang C, Wang G. Integrated Analysis of Methylomic and Transcriptomic Data to Identify Potential Diagnostic Biomarkers for Major Depressive Disorder. Genes. 2021; 12(2):178. https://doi.org/10.3390/genes12020178
Chicago/Turabian StyleXie, Yinping, Ling Xiao, Lijuan Chen, Yage Zheng, Caixia Zhang, and Gaohua Wang. 2021. "Integrated Analysis of Methylomic and Transcriptomic Data to Identify Potential Diagnostic Biomarkers for Major Depressive Disorder" Genes 12, no. 2: 178. https://doi.org/10.3390/genes12020178
APA StyleXie, Y., Xiao, L., Chen, L., Zheng, Y., Zhang, C., & Wang, G. (2021). Integrated Analysis of Methylomic and Transcriptomic Data to Identify Potential Diagnostic Biomarkers for Major Depressive Disorder. Genes, 12(2), 178. https://doi.org/10.3390/genes12020178