
Transcriptional Differences between Canine Cutaneous Epitheliotropic Lymphoma and Immune-Mediated Dermatoses

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Study Cohort
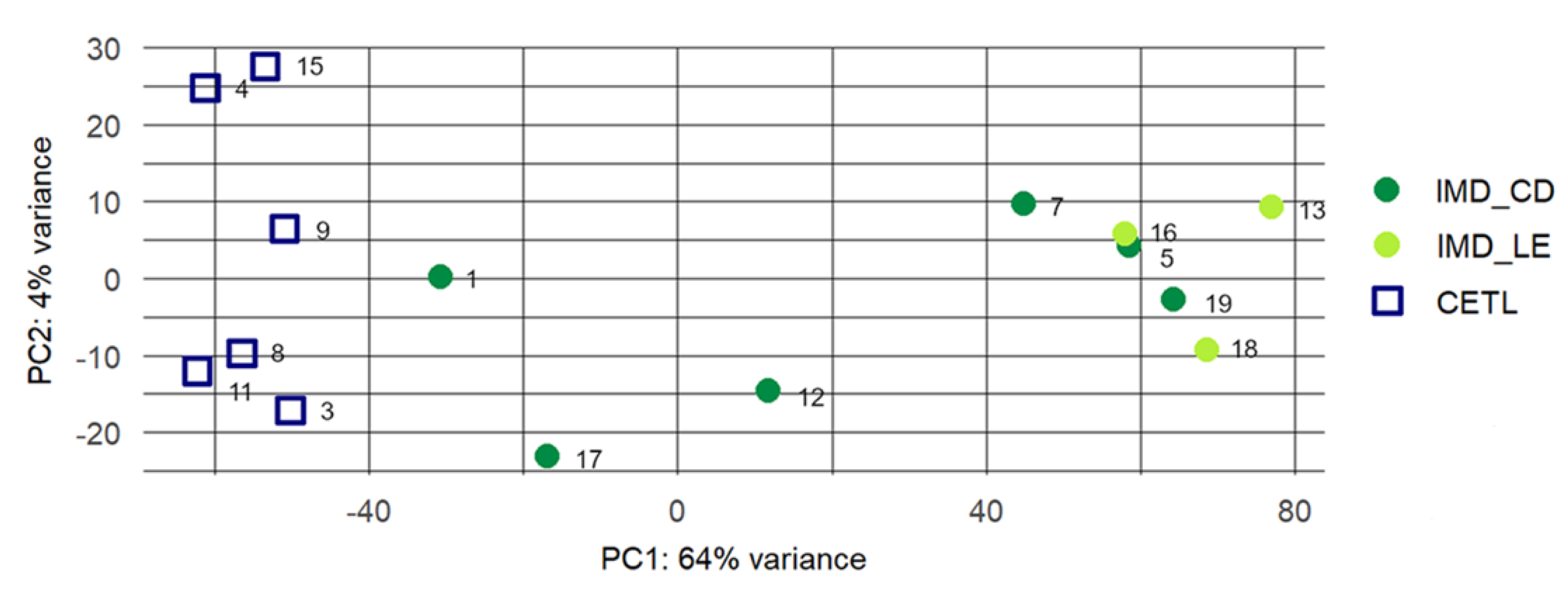
3.2. RNA Sequencing and Raw Data Analysis
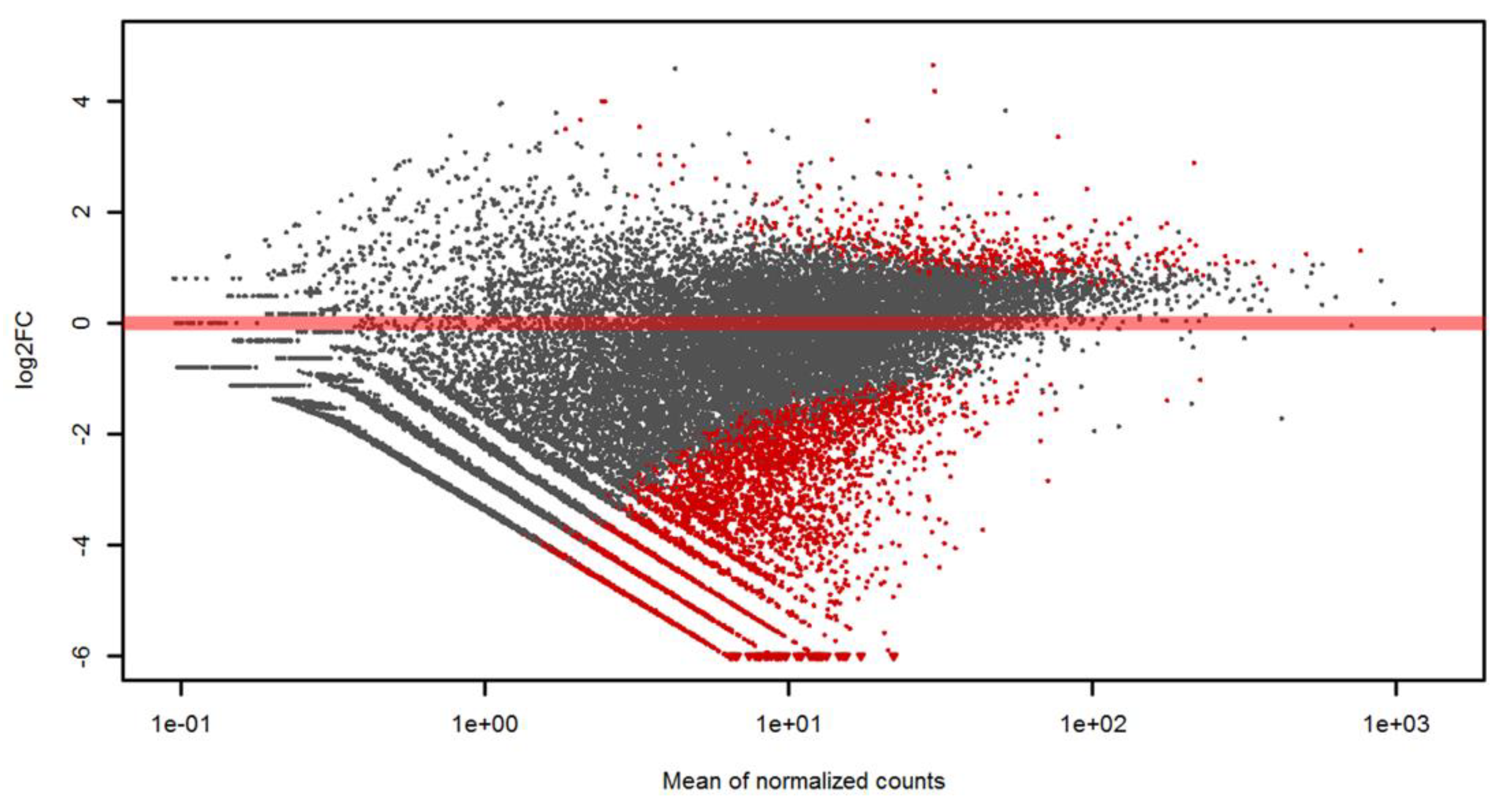
3.3. Differential Gene Expression Analysis
3.4. Pathway and Gene Enrichment Analysis
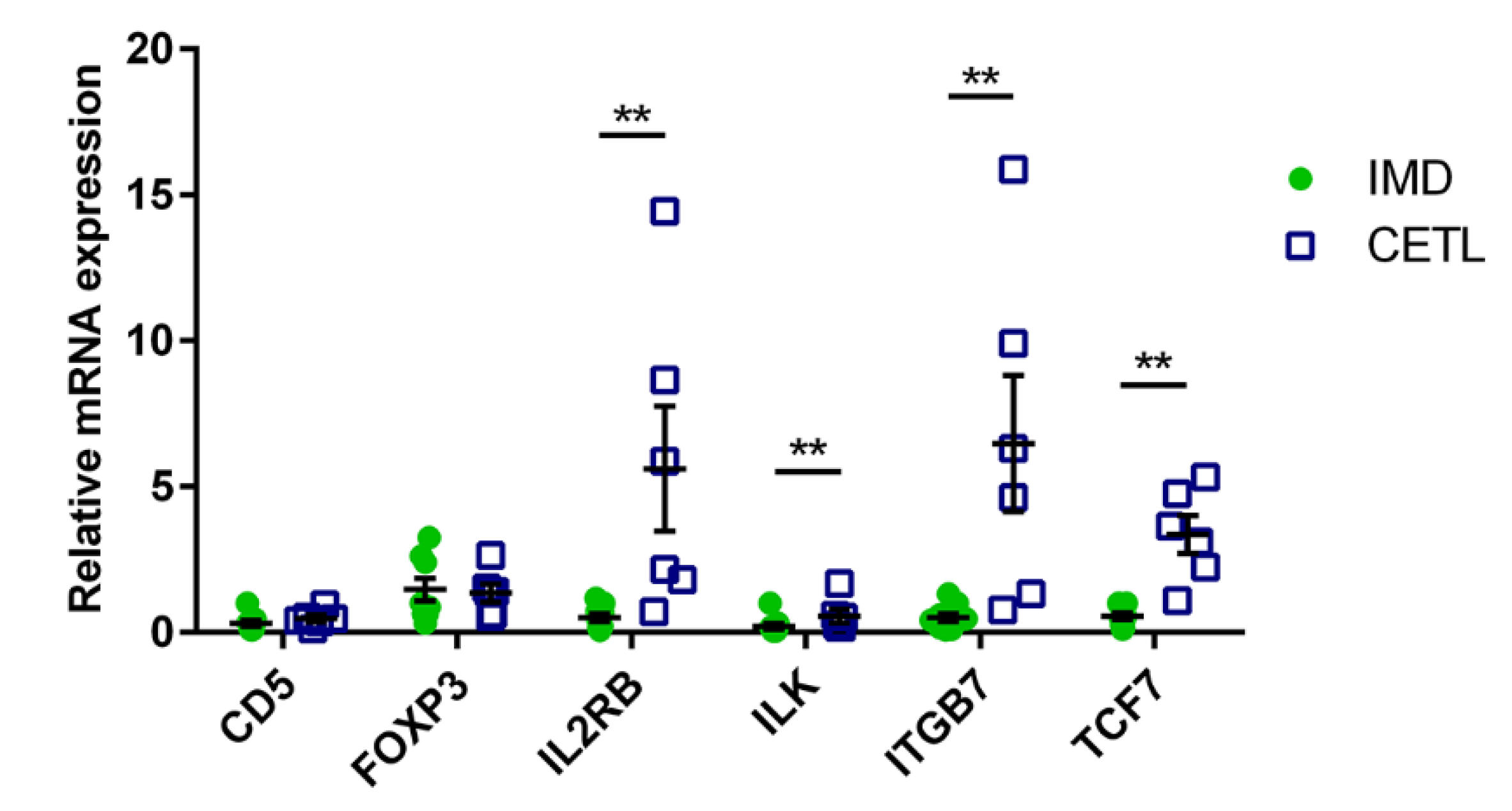
3.5. RT-qPCR
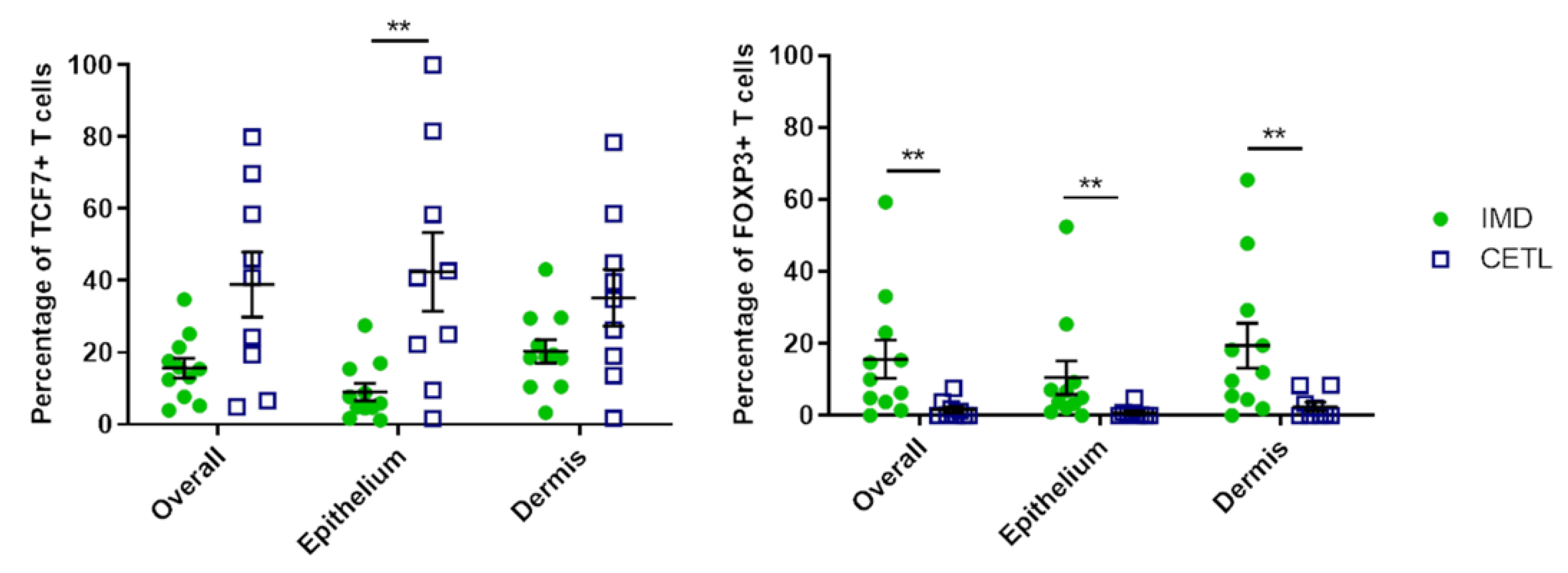
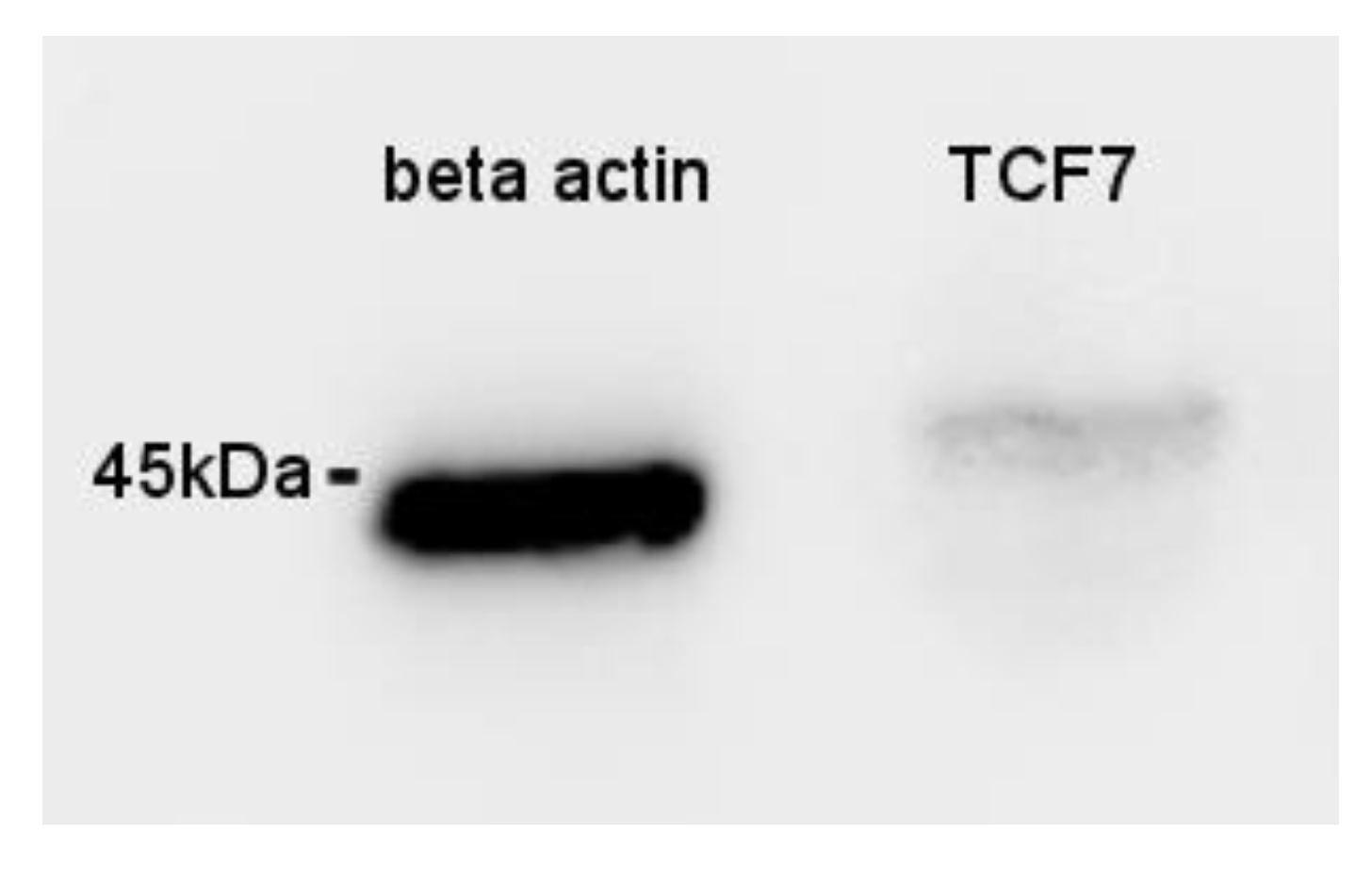
3.6. Immunohistochemistry
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Gross, T.L.; Ihrke, P.J.; Walder, E.J.; Affolter, V.K. (Eds.) Skin Disease of the Dog and Cat: Clinical and Histopathological Diagnosis, 2nd ed.; Blackwell Publishing Ltd.: Ames, IA, USA, 2005. [Google Scholar]
- Fontaine, J.; Heimann, M.; Day, M.J. Canine cutaneous epitheliotropic T-cell lymphoma: A review of 30 cases. Vet. Dermatol. 2010, 21, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Graf, R.; Pospischil, A.; Guscetti, F.; Meier, D.; Welle, M.; Dettwiler, M. Cutaneous Tumors in Swiss Dogs: Retrospective Data From the Swiss Canine Cancer Registry, 2008–2013. Vet. Pathol. 2018, 55, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Olivry, T.; Linder, K.E.; Banovic, F. Cutaneous lupus erythematosus in dogs: A comprehensive review. BMC Vet. Res. 2018, 14, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.; Griffin, C.; Campbell, K. Autoimmune and immune-mediated dermatoses. In Muller and Kirk’s Small Animal Dermatology; Saunders, Elsevier: St. Louis, MO, USA, 2013; pp. 432–500. [Google Scholar]
- Yager, J.A. Erythema multiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis: A comparative review. Vet. Dermatol. 2014, 25, 406-e64. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.F.; Affolter, V.K.; Graham, P.S.; Hirt, B. Canine epitheliotropic cutaneous T-cell lymphoma: An investigation of T-cell receptor immunophenotype, lesion topography and molecular clonality. Vet. Dermatol. 2009, 20, 569–576. [Google Scholar] [CrossRef]
- Chan, C.M.; Frimberger, A.E.; Moore, A.S. Clinical outcome and prognosis of dogs with histopathological features consistent with epitheliotropic lymphoma: A retrospective study of 148 cases (2003–2015) in Australia. Vet. Dermatol. 2018, 29, 154-e59. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.W.; Miller, W.H. Erythema multiforme in dogs and cats: Literature review and case material from the Cornell University College of Veterinary Medicine (1988–1996). Vet. Dermatol. 1999, 10, 297–309. [Google Scholar] [CrossRef]
- Keller, S.M.; Vernau, W.; Moore, P.F. Clonality Testing in Veterinary Medicine: A Review with Diagnostic Guidelines. Vet. Pathol. 2016, 53, 711–725. [Google Scholar] [CrossRef]
- Pimpinelli, N.; Olsen, E.A.; Santucci, M.; Vonderheid, E.; Haeffner, A.C.; Stevens, S.; Burg, G.; Cerroni, L.; Dreno, B.; Glusac, E.; et al. Defining early mycosis fungoides. J. Am. Acad. Dermatol. 2005, 53, 1053–1063. [Google Scholar] [CrossRef]
- Brachelente, C.; Cappelli, K.; Capomaccio, S.; Porcellato, I.; Silvestri, S.; Bongiovanni, L.; De Maria, R.; Verini Supplizi, A.; Mechelli, L.; Sforna, M. Transcriptome Analysis of Canine Cutaneous Melanoma and Melanocytoma Reveals a Modulation of Genes Regulating Extracellular Matrix Metabolism and Cell Cycle. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Grenier, J.K.; Foureman, P.A.; Sloma, E.A.; Miller, A.D. RNA-seq transcriptome analysis of formalin fixed, paraffin-embedded canine meningioma. PLoS ONE 2017, 12, e0187150. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.M.; Moore, P.F. A novel clonality assay for the assessment of canine T cell proliferations. Vet. Immunol. Immunopathol. 2012, 145, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC—A Quality Control Application for FastQ Files; Babraham Bioinformatics: Babraham, UK, 2010; Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 25 January 2021).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Wu, J.; Mao, X.; Cai, T.; Luo, J.; Wei, L. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34, W720–W724. [Google Scholar] [CrossRef]
- Hong, G.; Zhang, W.; Li, H.; Shen, X.; Guo, Z. Separate enrichment analysis of pathways for up- And downregulated genes. J. R. Soc. Interface 2014, 11, 20130950. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Yu, R.; Huang, Y.; Su, M.; Xiao, C.; Martinka, M.; Dutz, J.P.; Zhang, X.; Zheng, Z.; et al. Molecular Markers of Early-Stage Mycosis Fungoides. J. Investig. Dermatol. 2012, 132, 1698–1706. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Jones, D.A.; Sasseville, D.; Kupper, T.S. Transcriptional Profiles Predict Disease Outcome in Patients with Cutaneous T-Cell Lymphoma. Clin. Cancer Res. 2010, 16, 2106–2114. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Netchiporouk, E.; Cordeiro, B.; Dore, M.A.; Moreau, L.; Pehr, K.; Gilbert, M.; Zhou, Y.; Sasseville, D.; Kupper, T.S. The Use of Transcriptional Profiling to Improve Personalized Diagnosis and Management of Cutaneous T-Cell Lymphoma (CTCL). Clin. Cancer Res. 2015, 21, 2820–2829. [Google Scholar] [CrossRef]
- Shin, J.; Monti, S.; Aires, D.J.; Duvic, M.; Golub, T.; Jones, D.A.; Kupper, T.S. Lesional gene expression profiling in cutaneous T-cell lymphoma reveals natural clusters associated with disease outcome. Blood 2007, 110, 3015–3027. [Google Scholar] [CrossRef] [PubMed]
- Tracey, L.; Villuendas, R.; Dotor, A.M.; Spiteri, I.; Ortiz, P.; García, J.F.; Rodríguez Peralto, J.L.; Lawler, M.; Piris, M.A. Mycosis fungoides shows concurrent deregulation of multiple genes involved in the TNF signaling pathway: An expression profile study. Blood 2003, 102, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Troussard, A.A.; McDonald, P.C.; Wederell, E.D.; Mawji, N.M.; Filipenko, N.R.; Gelmon, K.A.; Kucab, J.E.; Dunn, S.E.; Emerman, J.T.; Bally, M.B.; et al. Preferential Dependence of Breast Cancer Cells versus Normal Cells on Integrin-Linked Kinase for Protein Kinase B/Akt Activation and Cell Survival. Cancer Res. 2006, 66, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Jin, L.; Tsutsumi-Ishii, Y.; Xu, Y.; McQueen, T.; Priebe, W.; Mills, G.B.; Ohsaka, A.; Nagaoka, I.; Andreeff, M.; et al. Activation of Integrin-Linked Kinase Is a Critical Prosurvival Pathway Induced in Leukemic Cells by Bone Marrow-Derived Stromal Cells. Cancer Res. 2007, 67, 684–694. [Google Scholar] [CrossRef]
- Koul, D.; Shen, R.; Bergh, S.; Lu, Y.; de Groot, J.F.; Liu, T.J.; Mills, G.B.; Yung, W.K.A. Targeting integrin-linked kinase inhibits Akt signaling pathways and decreases tumor progression of human glioblastoma. Mol. Cancer Ther. 2005, 4, 1681–1688. [Google Scholar] [CrossRef]
- Wada, D.A.; Wilcox, R.A.; Weenig, R.H.; Gibson, L.E. Paucity of intraepidermal FoxP3-positive T cells in cutaneous T-cell lymphoma in contrast with spongiotic and lichenoid dermatitis. J. Cutan. Pathol. 2010, 37, 535–541. [Google Scholar] [CrossRef]
- Fried, I.; Cerroni, L. FOXP3 in Sequential Biopsies of Progressive Mycosis Fungoides. Am. J. Dermatopathol. 2012, 34, 263–265. [Google Scholar] [CrossRef]
- Iwai, S.; Sueki, H.; Watanabe, H.; Sasaki, Y.; Suzuki, T.; Iijima, M. Distinguishing between erythema multiforme major and Stevens-Johnson syndrome/toxic epidermal necrolysis immunopathologically. J. Dermatol. 2012, 39, 781–786. [Google Scholar] [CrossRef]
- Solomon, G.J.; Magro, C.M. Foxp3 expression in cutaneous T-cell lymphocytic infiltrates. J. Cutan. Pathol. 2008, 35, 1032–1039. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Maccoux, L.J.; Clements, D.N.; Salway, F.; Day, P.J.R. Identification of new reference genes for the normalisation of canine osteoarthritic joint tissue transcripts from microarray data. BMC Mol. Biol. 2007, 8, 62. [Google Scholar] [CrossRef]
- Peters, I.R.; Peeters, D.; Helps, C.R.; Day, M.J. Development and application of multiple internal reference (housekeeper) gene assays for accurate normalisation of canine gene expression studies. Vet. Immunol. Immunopathol. 2007, 117, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Schlotter, Y.M.; Veenhof, E.Z.; Brinkhof, B.; Rutten, V.P.M.G.; Spee, B.; Willemse, T.; Penning, L.C. A GeNorm algorithm-based selection of reference genes for quantitative real-time PCR in skin biopsies of healthy dogs and dogs with atopic dermatitis. Vet. Immunol. Immunopathol. 2009, 129, 115–118. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, I.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, R. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 0034.1–0034.11. [Google Scholar] [CrossRef]
- Boozer, L.B.; Davis, T.W.; Borst, L.B.; Zseltvay, K.M.; Olby, N.J.; Mariani, C.L. Characterization of Immune Cell Infiltration Into Canine Intracranial Meningiomas. Vet. Pathol. 2012, 49, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Mao-De, L.; Jing, X. Ribosomal Proteins and Colorectal Cancer. Curr. Genomics 2007, 8, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Henry, J.L.; Coggin, D.L.; King, C.R. High-Level Expression of the Ribosomal Protein L19 in Human Breast Tumors That Overexpress erbB-21. Cancer Res. 1993, 53, 1403–1408. [Google Scholar] [PubMed]
- Vaarala, M.H.; Porvari, K.S.; Kyllönen, A.P.; Mustonen, M.V.J.; Lukkarinen, O.; Vihko, P.T. Several genes encoding ribosomal proteins are over-expressed in prostate-cancer cell lines: Confirmation of L7a and L37 over-expression in prostate-cancer tissue samples. Int. J. Cancer 1998, 78, 27–32. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, C.; Zhou, J.; Wang, X.; Wu, M.; Liu, Z. Cloning and characterization of full-length human ribosomal protein L15 cDNA which was overexpressed in esophageal cancer. Gene 2001, 263, 205–209. [Google Scholar] [CrossRef]
- Kim, J.H.; You, K.R.; Kim, I.H.; Cho, B.H.; Kim, C.Y.; Kim, D.G. Over-Expression of the Ribosomal Protein L36a Gene Is Associated with Cellular Proliferation in Hepatocellular Carcinoma. Hepatology 2004, 39, 129–138. [Google Scholar] [CrossRef]
- Montanaro, L.; Treré, D.; Derenzini, M. Nucleolus, ribosomes, and cancer. Am. J. Pathol. 2008, 173, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Petit, V.; Thiery, J.-P. Focal adhesions: Structure and dynamics. Biol. Cell 2000, 92, 477–494. [Google Scholar] [CrossRef]
- Berman, A.E.; Kozlova, N.I.; Morozevich, G.E. Integrins: Structure and Signaling. Biochemistry 2003, 68, 1284–1299. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lonsdorf, A.; Hwang, S.T. Cutaneous T Cell Lymphoma: Roles for Chemokines and Chemokine Receptors. J. Investig. Dermatol 2009, 129, 1115–1119. [Google Scholar] [CrossRef][Green Version]
- Geppetti, P.; Veldhuis, N.A.; Lieu, T.M.; Bunnett, N.W. G Protein-Coupled Receptors: Dynamic Machines for Signaling Pain and Itch. Neuron 2015, 88, 635–649. [Google Scholar] [CrossRef]
- Chimura, N.; Kondo, N.; Shibata, S.; Kimura, T.; Mori, T.; Hoshino, Y.; Murayama, N.; Nagata, M.; Ide, K.; Nishifuji, K.; et al. Gene transcription analysis in lesional skin of canine epitheliotropic cutaneous lymphoma using quantitative real-time RT-PCR. Vet. Immunol. Immunopathol. 2011, 144, 329–336. [Google Scholar] [CrossRef]
- Chimura, N.; Iio, A.; Ozaki, E.; Mori, T.; Ito, Y.; Murayama, N.; Nagata, M.; Ide, K.; Nishifuji, K.; Kamishina, H.; et al. Transcription profile of chemokine receptors, cytokines and cytotoxic markers in peripheral blood of dogs with epitheliotropic cutaneous lymphoma. Vet. Dermatol. 2013, 24. [Google Scholar] [CrossRef]
- Van de Wetering, M.; de Lau, W.; Clevers, H. WNT Signaling and Lymphocyte Development. Cell 2002, 109, S13–S19. [Google Scholar] [CrossRef]
- Dorfman, D.M.; Greisman, H.A.; Shahsafaei, A. Loss of Expression of the WNT/β-Catenin-Signaling Pathway Transcription Factors Lymphoid Enhancer Factor-1 (LEF-1) and T Cell Factor-1 (TCF-1) in a Subset of Peripheral T Cell Lymphomas. Am. J. Pathol. 2003, 162, 1539–1544. [Google Scholar] [CrossRef]
- Leonard, W.J.; Krönke, M.; Peffer, N.J.; Depper, J.M.; Greene, W.C. Interleukin 2 receptor gene expression in normal human T lymphocytes. Proc. Natl. Acad. Sci. USA 1985, 82, 6281–6285. [Google Scholar] [CrossRef]
- Eriksen, K.; Kaltoft, K.; Mikkelsen, G.; Nielsen, M.; Zhang, Q.; Geisler, C.; Nissen, M.; Röpke, C.; Wasik, M.; Ødum, N. Constitutive STAT3-activation in Sezary syndrome: Tyrphostin AG490 inhibits STAT3-activation, interleukin-2 receptor expression and growth of leukemic Sezary cells. Leukemia 2001, 15, 787–793. [Google Scholar] [CrossRef]
- Van Der Fits, L.; Out-Luiting, J.J.; Van Leeuwen, M.A.; Samsom, J.N.; Willemze, R.; Tensen, C.P.; Vermeer, M.H. Autocrine IL-21 Stimulation Is Involved in the Maintenance of Constitutive STAT3 Activation in Sézary Syndrome. J. Investig. Dermatol. 2012, 132, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Leonard, W.J. Signaling from the IL-2 Receptor to the Nucleus. Cytokine Growth Factor Rev. 1997, 8, 313–332. [Google Scholar] [CrossRef]
- Simonitsch, I.; Volc-Platzer, B.; Mosberger, I.; Radaszkiewicz, T. Expression of Monoclonal Antibody HML-1-Defined alpha E beta 7 Integrin in Cutaneous T Cell Lymphoma. Am. J. Pathol. 1994, 145, 1148–1158. [Google Scholar]
- Oloumi, A.; Syam, S.; Dedhar, S. Modulation of Wnt3a-mediated nuclear β-catenin accumulation and activation by integrin-linked kinase in mammalian cells. Oncogene 2006, 25, 7747–7757. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Sinha, S.; Williams, C.; Cyrille, M.; Heller, E.; Snapper, S.B.; Georgopoulos, K.; St-Arnaud, R.; Force, T.; Dedhar, S.; et al. Targeted Deletion of Integrin-Linked Kinase Reveals a Role in T-Cell Chemotaxis and Survival. Mol. Cell. Biol. 2005, 25, 11145–11155. [Google Scholar] [CrossRef]
- Bossel Ben-Moshe, N.; Gilad, S.; Perry, G.; Benjamin, S.; Balint-Lahat, N.; Pavlovsky, A.; Halperin, S.; Markus, B.; Yosepovich, A.; Barshack, I.; et al. mRNA-seq whole transcriptome profiling of fresh frozen versus archived fixed tissues. BMC Genomics 2018, 19, 419. [Google Scholar] [CrossRef]
- Esteve-Codina, A.; Arpi, O.; Martinez-García, M.; Pineda, E.; Mallo, M.; Gut, M.; Carrato, C.; Rovira, A.; Lopez, R.; Tortosa, A.; et al. A Comparison of RNA-Seq Results from Paired Formalin-Fixed Paraffin-Embedded and Fresh-Frozen Glioblastoma Tissue Samples. PLoS ONE 2017, 12, e0170632. [Google Scholar] [CrossRef]
- Leek, J.T.; Scharpf, R.B.; Bravo, H.C.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession | Forward | Reverse | Product Size (bp) |
|---|---|---|---|---|
| CCZ1 | XM_536878.6 | GCAGGAAGGGATTCTCCAG | GGTCCAGTAAGAAATCTTCCATAA | 74 |
| GUSβ | NM_001003191.1 | GTGCTGGATCAGAAACGCAA | CTTTGGGTTGTCTCTGGCGA | 136 |
| RPL8 | XM_532360 | CCATGAATCCTGTGGAGC | GTAGAGGGTTTGCCGATG | 64 |
| RPL32 | XM_848016 | TGGTTACAGGAGCAACAAGAAA | GCACATCAGCAGCACTTCA | 100 |
| RPS5 | XM_533568 | TCACTGGTGAGAACCCCCT | CCTGATTCACACGGCGTAG | 141 |
| RPS18 | XM_532106 | TGCTCATGTGGTATTGAGGAA | TCTTATACTGGCGTGGATTCTG | 116 |
| RPS19 | XM_533657 | CCTTCCTCAAAAAGTCTGGG | GTTCTCATCGTAGGGAGCAAG | 95 |
| CD5 | XM_022405839.1 | CTTAGGCTGGCCTTGAAGCT | ACACTGGTGTTGCAGTTGGA | 143 |
| FOXP3 | NM_001168461.1 | AAATTCCACAACATGCGCCC | AGGCAAACATGCGTGTGAAC | 124 |
| IL2RB | NM_001286851.1 | TCCTGTGAGCTGCTCCCTAT | ATCCTCCACCTCTCCCCTTC | 137 |
| ILK | XM_022407778.1 | CACGGTTAGGGGAGTGTGTC | CCGTGTGGCAAGTGACAAAG | 163 |
| ITGB7 | XM_022411473.1 | GACTCCAGCAACGTGGTACA | CCCTCTTCTCAGGATCCCCA | 136 |
| TCF7 | XM_022425289.1 | GCAGAGACTTTTCCCCGACA | GCATGAGCAGATTGAAGGCG | 116 |
| Case No. | Disease Type (with Precise IMD Diagnosis) | Breed | Sex 1 | Age at Time of Diagnosis (Years and Months) | Overall Survival Time (Days) | PARR 2 | Tests Applied |
|---|---|---|---|---|---|---|---|
| 1 | IMD (PCD) | Jack Russel Terrier | M | 10 y 10 m | 78 | polyclonal | RNA-seq, RT-qPCR, IHC |
| 2 | CETL | Mixed | F | 7 y 9 m | 166 | clonal | IHC |
| 3 | CETL | Cocker Spaniel | MC | 7 y 7 m | 230 | clonal | RNA-seq, RT-qPCR, IHC |
| 4 | CETL | Pyrenean Shepherd | FS | 12 y | 277 | clonal | RNA-seq, RT-qPCR, IHC |
| 5 | IMD (PCD) | Briard | M | 10 y 2 m | 1324 | polyclonal | RNA-seq, RT-qPCR, IHC |
| 6 | CETL | West Highland White Terrier | FS | 11 y 8 m | 187 | negative | IHC |
| 7 | IMD (PCD) | American Staffordshire Terrier | MC | 11 y 8 m | 54 | polyclonal | RNA-seq, RT-qPCR, IHC |
| 8 | CETL | Cocker Spaniel | FS | 13 y 9 m | 179 | clonal | RNA-seq, RT-qPCR, IHC |
| 9 | CETL | Mixed | MC | 11 y 3 m | 18 | clonal | RNA-seq, RT-qPCR, IHC |
| 10 | CETL | Cocker Spaniel | FS | 11 y 1 m | 6 | clonal | IHC |
| 11 | CETL | Golden Retriever | F | 9 y 2 m | 57 | clonal | RNA-seq, RT-qPCR, IHC |
| 12 | IMD (PCD) | Shetland Sheepdog | F | 10 y 7 m | alive | polyclonal | RNA-seq, RT-qPCR, IHC |
| 13 | IMD (LE) | Border Collie | MC | 5 y 3 m | alive | negative | RNA-seq, RT-qPCR, IHC |
| 14 | IMD (PCD) | Podenco Canario | FS | 9 y 8 m | 45 | polyclonal | IHC |
| 15 | CETL | Boxer | M | 8 y 8 m | 134 | clonal | RNA-seq, RT-qPCR, IHC |
| 16 | IMD (LE) | Magyar Vizsla | F | 2 y 3 m | 2 | polyclonal | RNA-seq, RT-qPCR, IHC |
| 17 | IMD (PCD) | Great Pyrenees | FS | 5 y 11 m | alive | polyclonal | RNA-seq, RT-qPCR, IHC |
| 18 | IMD (LE) | Rhodesian Ridgeback | M | 2 y 6 m | alive | polyclonal | RNA-seq, RT-qPCR, IHC |
| 19 | IMD (PCD) | Yorkshire Terrier | FS | 11 y 2 m | alive | polyclonal | RNA-seq, RT-qPCR, IHC |
| 20 | IMD (LE) | Tervuren | M | 2 y 3 m | alive | polyclonal | IHC |
| Pathway | Enrichment Analysis Tool | Database | p-Value |
|---|---|---|---|
| EIF2 signaling | IPA | IPA | 1.66 × 10−7 |
| ILK signaling | IPA | IPA | 1.02 × 10−6 |
| Integrin signaling | IPA | IPA | 9.55 × 10−5 |
| Ribosome | KOBAS | KEGG | 1.87 × 10−6 |
| Cytosolic ribosome | KOBAS | GO | 1.87 × 10−6 |
| Ribosomal subunit | KOBAS | GO | 1.87 × 10−6 |
| Focal adhesion | KOBAS | GO | 1.87 × 10−6 |
| Pathway | Enrichment Analysis Tool | Database | p-Value |
|---|---|---|---|
| GABA receptor signaling | IPA | IPA | 5.01 × 10−12 |
| Cellular effects of sildenafil (Viagra) | IPA | IPA | 8.51 × 10−9 |
| Glutamate receptor signaling | IPA | IPA | 8.51 × 10−8 |
| Olfactory transduction | KOBAS | KEGG | 2.01 × 10−43 |
| Neuroactive ligand–receptor interaction | KOBAS | KEGG | 2.70 × 10−5 |
| Nicotine addiction | KOBAS | KEGG | 1.39 × 10−3 |
| Transmembrane signaling receptor activity | KOBAS | GO | 2.86 × 10−9 |
| Signaling receptor activity | KOBAS | GO | 2.86 × 10−9 |
| G-protein coupled receptor activity | KOBAS | GO | 2.86 × 10−9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerber, N.; Brunner, M.A.T.; Jagannathan, V.; Leeb, T.; Gerhards, N.M.; Welle, M.M.; Dettwiler, M. Transcriptional Differences between Canine Cutaneous Epitheliotropic Lymphoma and Immune-Mediated Dermatoses. Genes 2021, 12, 160. https://doi.org/10.3390/genes12020160
Gerber N, Brunner MAT, Jagannathan V, Leeb T, Gerhards NM, Welle MM, Dettwiler M. Transcriptional Differences between Canine Cutaneous Epitheliotropic Lymphoma and Immune-Mediated Dermatoses. Genes. 2021; 12(2):160. https://doi.org/10.3390/genes12020160
Chicago/Turabian StyleGerber, Nadja, Magdalena A. T. Brunner, Vidhya Jagannathan, Tosso Leeb, Nora M. Gerhards, Monika M. Welle, and Martina Dettwiler. 2021. "Transcriptional Differences between Canine Cutaneous Epitheliotropic Lymphoma and Immune-Mediated Dermatoses" Genes 12, no. 2: 160. https://doi.org/10.3390/genes12020160
APA StyleGerber, N., Brunner, M. A. T., Jagannathan, V., Leeb, T., Gerhards, N. M., Welle, M. M., & Dettwiler, M. (2021). Transcriptional Differences between Canine Cutaneous Epitheliotropic Lymphoma and Immune-Mediated Dermatoses. Genes, 12(2), 160. https://doi.org/10.3390/genes12020160