
Ancient DNA from the Asiatic Wild Dog (Cuon alpinus) from Europe

 , , ,
, , ,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Description
2.2. Mitochondrial DNA Sequencing
2.2.1. Modern Sample
2.2.2. Ancient Samples
2.2.3. Capture
2.2.4. Sequencing
2.3. Data Processing
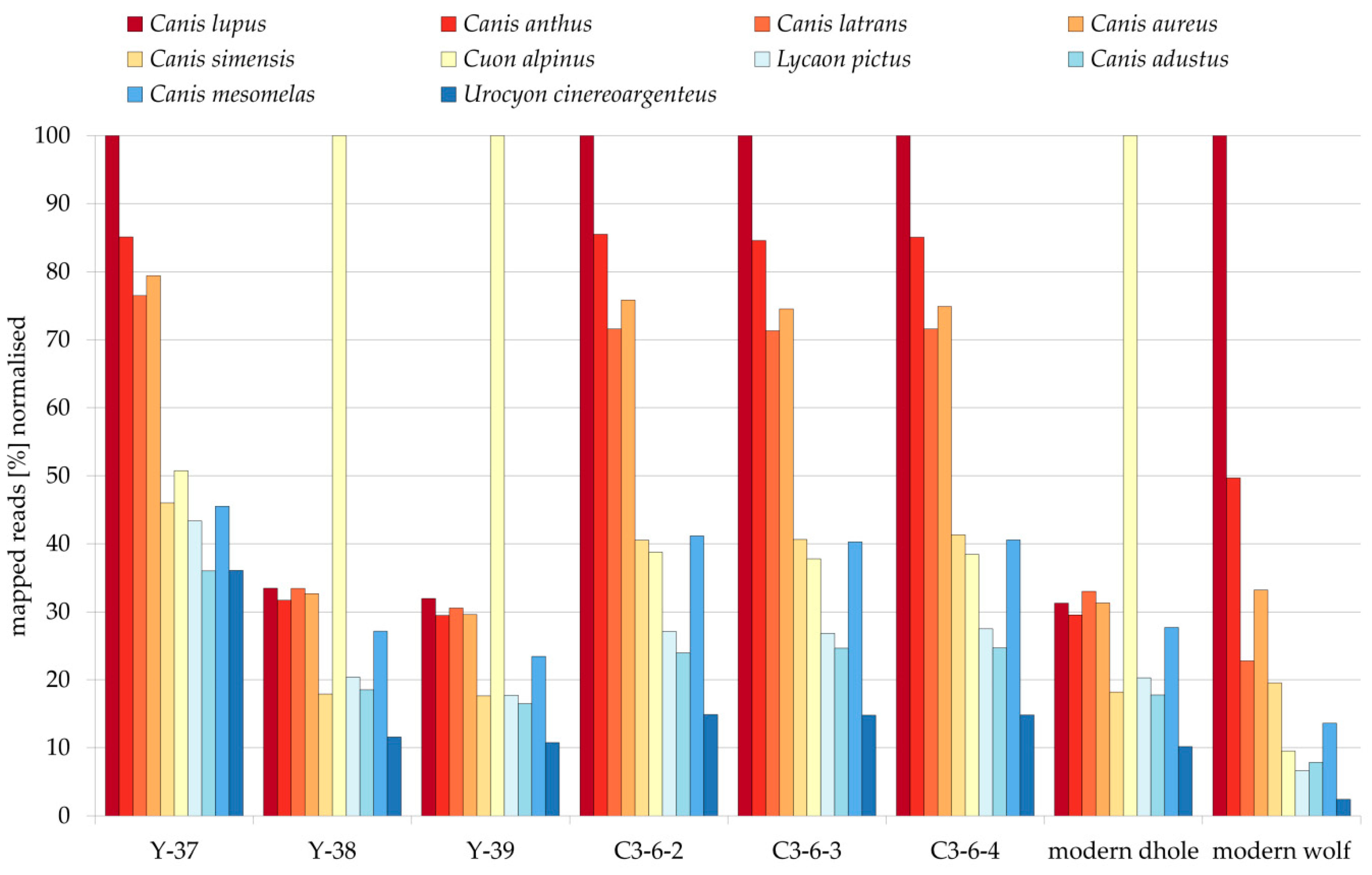
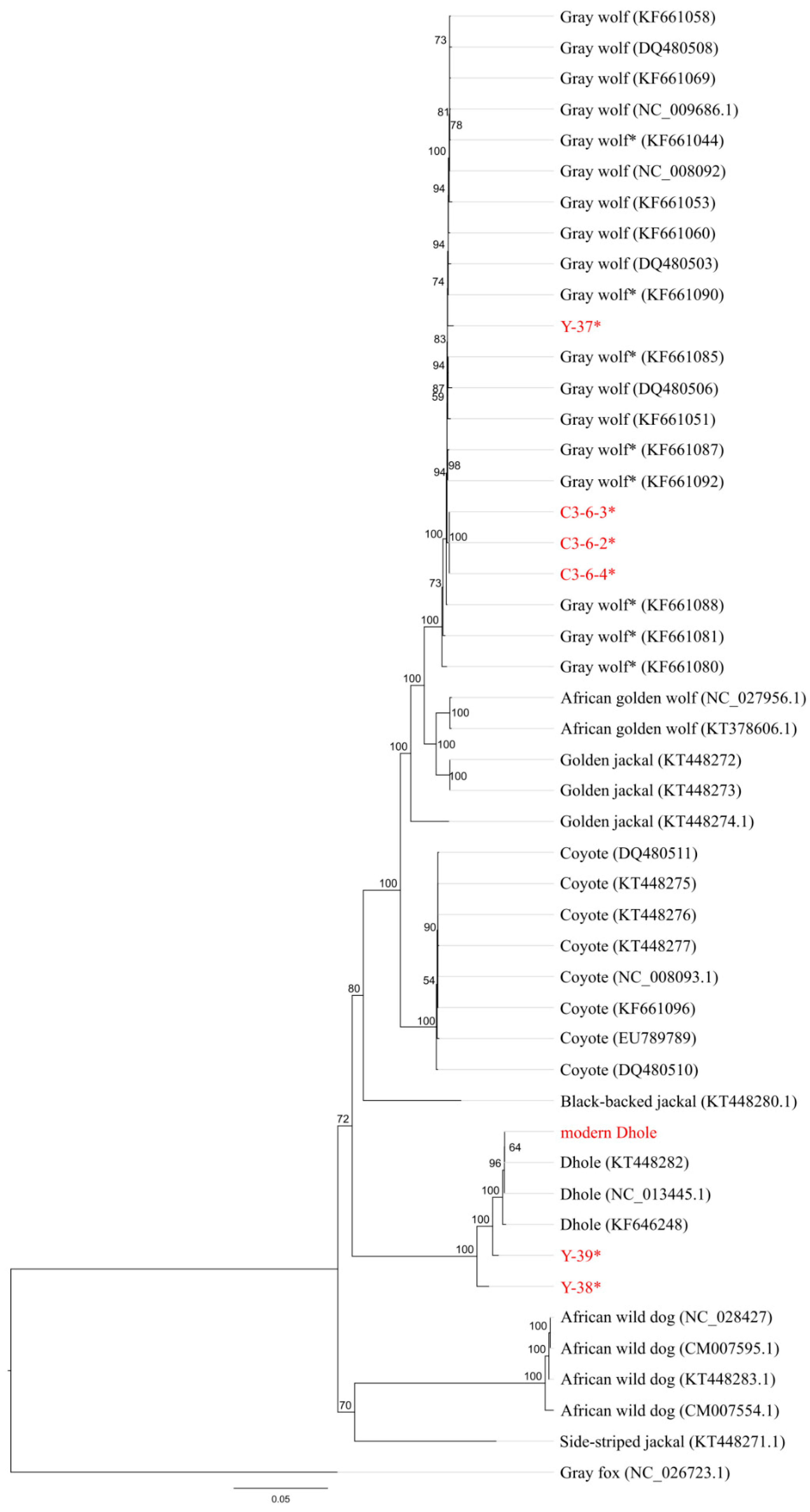
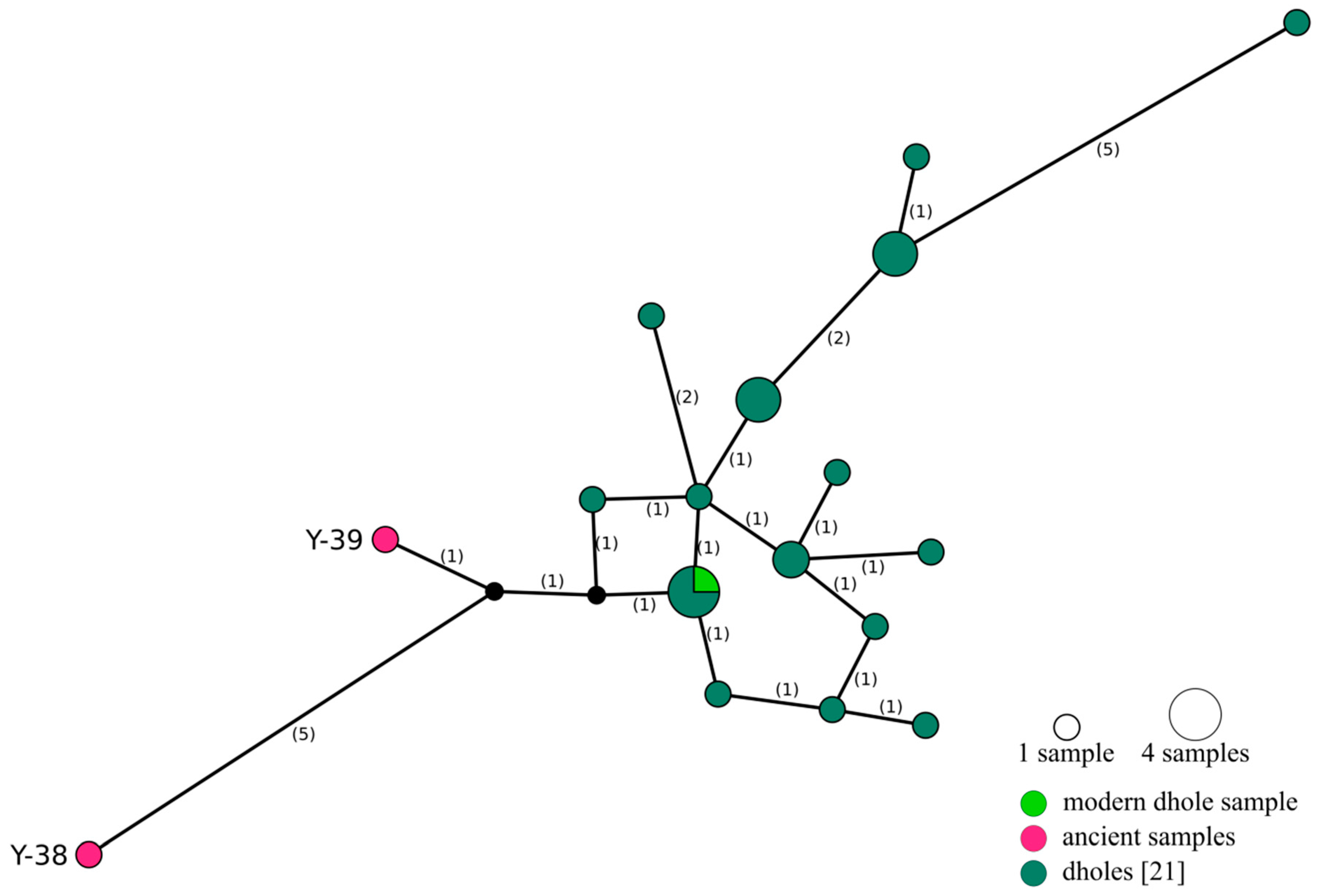
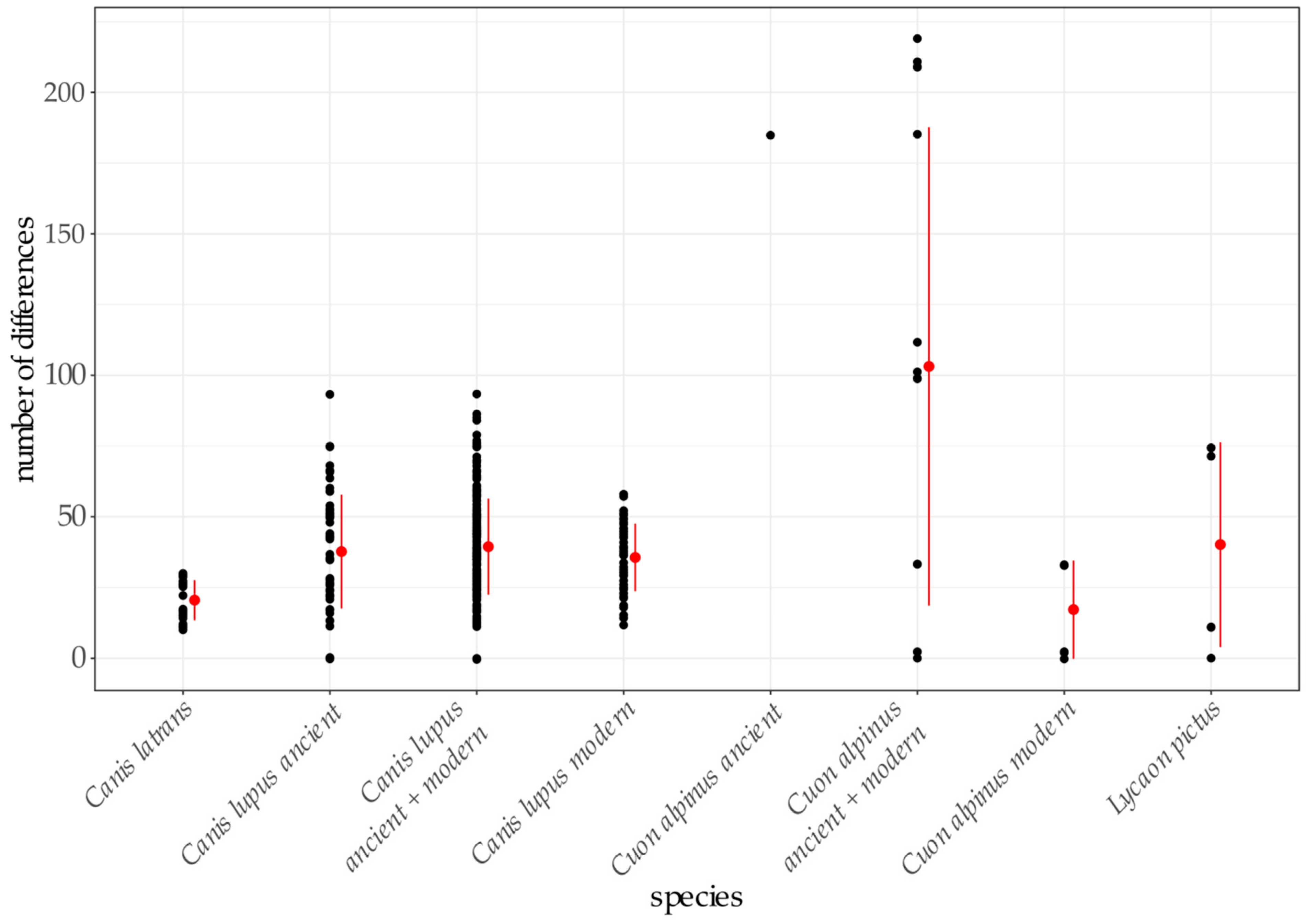
3. Results
4. Discussion
4.1. Palaeogenetic Identification of Fossils
4.2. (Mis) Identification of Dhole Remains
4.3. Relationship between Ancient and Modern Dholes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, O.J.F. Tasmanian Devil (Sarcophilus harrisii) Extinction on the Australian Mainland in the Mid-Holocene: Multicausality and ENSO Intensification. Alcheringa Australas. J. Palaeontol. 2006, 30, 49–57. [Google Scholar] [CrossRef]
- Paijmans, J.L.A.; Barlow, A.; Förster, D.W.; Henneberger, K.; Meyer, M.; Nickel, B.; Nagel, D.; Worsøe Havmøller, R.; Baryshnikov, G.F.; Joger, U.; et al. Historical Biogeography of the Leopard (Panthera pardus) and Its Extinct Eurasian Populations. BMC Evol. Biol. 2018, 18, 156. [Google Scholar] [CrossRef] [PubMed]
- Kurtén, B. Pleistocene Mammals of Europe; Weidenfeld and Nicolson: London, UK, 1968. [Google Scholar]
- Baryshnikov, G. The Dhole, Cuon alpinus (Carnivora, Canidae), from the Upper Pleistocene of the Caucasus. Acta Zool. Crac. 1996, 39, 67–73. [Google Scholar]
- Ghezzo, E.; Rook, L. Cuon alpinus (Pallas, 1811) (Mammalia, Carnivora) from Equi (Late Pleistocene, Massa—Carrara, Italy): Anatomical Analysis and Palaeoethological Contextualisation. Rend. Fis. Acc. Lincei 2014, 25, 491–504. [Google Scholar] [CrossRef]
- Kurtén, B.; Anderson, E. Pleistocene Mammals of North America; Columbia University Press: New York, NY, USA, 1980. [Google Scholar]
- Petrucci, M.; Romiti, S.; Sardella, R. The Middle-Late Pleistocene Cuon Hodgson, 1838 (Carnivora, Canidae) from Italy. Bollettino della Societa Paleontologica Italiana 2012, 51, 137–148. [Google Scholar] [CrossRef]
- Pérez Ripoll, M.; Morales Pérez, J.V.; Sanchis Serra, A.; Aura Tortosa, J.E.; Montañana, I.S. Presence of the Genus Cuon in Upper Pleistocene and Initial Holocene Sites of the Iberian Peninsula: New Remains Identified in Archaeological Contexts of the Mediterranean Region. J. Archaeol. Sci. 2010, 37, 437–450. [Google Scholar] [CrossRef]
- Tedford, R.H.; Wang, X.; Taylor, B.E. Phylogenetic Systematics of the North American Fossil Caninae (Carnivora: Canidae). Bull. Am. Mus. Nat. Hist. 2009, 1–218. [Google Scholar] [CrossRef]
- Thenius, E. Zur Abstammung Der Rotwölfe (Gattung Cuon HODGSON). Österr. Zool. Z. 1954, 5, 377–387. [Google Scholar]
- Wang, X.; Tedford, R.H. Dogs: Their Fossil Relatives and Evolutionary History; Columbia University Press: New York, NY, USA, 2008. [Google Scholar]
- Cohen, J.A. Cuon alpinus. Mamm. Species 1978, 1–3. [Google Scholar] [CrossRef]
- Durbin, L.S.; Venkataraman, A.; Hedges, S.; Duckworth, W. Dhole (Cuon alpinus). In Canids: Foxes, Wolves, Jackals and Dogs. Status Suvey and Conservation Action Plan; IUCN/SSC Action Plans for the Conservation of Biological Diversity; Hoffmann, M., Macdonald, D.W., Sillero-Zubiri, C., Eds.; IUCN: Gland, Switzerland; Cambridge, UK, 2004; pp. 210–219. ISBN 2-8317-0786-2. [Google Scholar]
- Kamler, J.F.; Songsasen, N.; Jenks, K.; Srivathsa, A.; Sheng, L.; Kunkel, K. Cuon alpinus; The IUCN Red List of Threatened Species 2015: E.T5953A72477893; IUCN: Gland, Switzerland, 2015. [Google Scholar]
- Gopalakrishnan, S.; Sinding, M.-H.S.; Ramos-Madrigal, J.; Niemann, J.; Samaniego Castruita, J.A.; Vieira, F.G.; Carøe, C.; Montero, M.d.M.; Kuderna, L.; Serres, A.; et al. Interspecific Gene Flow Shaped the Evolution of the Genus Canis. Curr. Biol. 2018, 28, 3441–3449.e5. [Google Scholar] [CrossRef]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J.; Zody, M.C.; et al. Genome Sequence, Comparative Analysis and Haplotype Structure of the Domestic Dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Wayne, R.K.; Geffen, E.; Girman, D.J.; Koepfli, K.P.; Lau, L.M.; Marshall, C.R. Molecular Systematics of the Canidae. Syst. Biol. 1997, 46, 622–653. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, L. The Complete Mitochondrial Genome of Dhole Cuon alpinus: Phylogenetic Analysis and Dating Evolutionary Divergence within Canidae. Mol. Biol. Rep. 2011, 38, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Bardeleben, C.; Moore, R.L.; Wayne, R.K. A Molecular Phylogeny of the Canidae Based on Six Nuclear Loci. Mol. Phylogenetics Evol. 2005, 37, 815–831. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, H.; Zhang, J.; Zhao, C.; Sha, W. The Complete Mitochondrial Genome of Cuon alpinus lepturus. Mitochondrial DNA 2015, 26, 767–768. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, A.; Babu, V.N.; Hedges, S.; Venkataraman, A.B.; Maclean, N.; Morin, P.A. Phylogeography, Genetic Structure, and Diversity in the Dhole (Cuon alpinus). Mol. Ecol. 2005, 14, 2281–2297. [Google Scholar] [CrossRef]
- Koepfli, K.-P.; Pollinger, J.; Godinho, R.; Robinson, J.; Lea, A.; Hendricks, S.; Schweizer, R.M.; Thalmann, O.; Silva, P.; Fan, Z.; et al. Genome-Wide Evidence Reveals That African and Eurasian Golden Jackals Are Distinct Species. Curr. Biol. 2015, 25, 2158–2165. [Google Scholar] [CrossRef]
- Sheldon, J.W. Wild Dogs: The Natural History of the Non-Domestic Canidae, 1st ed.; Academic Press Inc.: San Diego, CA, USA, 1992; ISBN 978-0-12-639375-0. [Google Scholar]
- Baryshnikov, G.F. Pleistocene Canidae (Mammalia, Carnivora) from the Paleolithic Kudaro Caves in the Caucasus. Russ. J. Theriol. 2012, 11, 71–120. [Google Scholar] [CrossRef]
- Stehlík, A. Cuon auropaeus Bourguignat z Plistocenních Usazenin Jeskyně Jáchymky v Josefském Údolí u Adamova. Práce Moravské Přírodověckě Společnosti 1944, 16, 1–21. [Google Scholar]
- Wiszniowska, T. Carnivora. In Excavation in the Bacho Kiro Cave (Bulgaria). Final Report; Państwowe Wydawnictwo Naukowe: Warszawa, Poland, 1982; pp. 52–55. [Google Scholar]
- Drăgușin, V.; Vasile, Ș.; Terhune, C.; Petculescu, A.; Robu, M.; Vlaicu, M.; Constantin, S.; Știucă, E.; Taron, U.H.; Hofreiter, M.; et al. Late Pleistocene Occurrence of Cuon alpinus in Romania. In Proceedings of the Eleventh Romanian Symposium on Palaeontology, Bucharest, Romania, 27–28 September 2017; Lazăr, I., Grădinaru, M., Vasile, Ș., Eds.; University of Bucharest: Bucharest, Romania; p. 27. [Google Scholar]
- Hublin, J.-J. The Modern Human Colonization of Western Eurasia: When and Where? Quat. Sci. Rev. 2015, 118, 194–210. [Google Scholar] [CrossRef]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an Enhanced Web Interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed]
- Henneberger, K.; Barlow, A.; Paijmans, J.L.A. Double-Stranded Library Preparation for Ancient and Other Degraded Samples. Methods Mol. Biol. 2019, 1963, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Dabney, J.; Knapp, M.; Glocke, I.; Gansauge, M.-T.; Weihmann, A.; Nickel, B.; Valdiosera, C.; García, N.; Pääbo, S.; Arsuaga, J.-L.; et al. Complete Mitochondrial Genome Sequence of a Middle Pleistocene Cave Bear Reconstructed from Ultrashort DNA Fragments. Proc. Natl. Acad. Sci. USA 2013, 110, 15758–15763. [Google Scholar] [CrossRef] [PubMed]
- Gansauge, M.-T.; Meyer, M. Single-Stranded DNA Library Preparation for the Sequencing of Ancient or Damaged DNA. Nat. Protoc. 2013, 8, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Korlević, P.; Gerber, T.; Gansauge, M.-T.; Hajdinjak, M.; Nagel, S.; Aximu-Petri, A.; Meyer, M. Reducing Microbial and Human Contamination in DNA Extractions from Ancient Bones and Teeth. BioTechniques 2015, 59, 87–93. [Google Scholar] [CrossRef]
- González Fortes, G.; Paijmans, J.L.A. Whole-Genome Capture of Ancient DNA Using Homemade Baits. In Ancient DNA: Methods and Protocols; Shapiro, B., Barlow, A., Heintzman, P.D., Hofreiter, M., Paijmans, J.L.A., Soares, A.E.R., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 93–105. ISBN 978-1-4939-9176-1. [Google Scholar]
- Paijmans, J.L.A.; Baleka, S.; Henneberger, K.; Taron, U.H.; Trinks, A.; Westbury, M.V.; Barlow, A. Sequencing Single-Stranded Libraries on the Illumina NextSeq 500 Platform. arXiv 2017, arXiv:1711.11004. [Google Scholar]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet.journal. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Freedman, A.H.; Gronau, I.; Schweizer, R.M.; Vecchyo, D.O.-D.; Han, E.; Silva, P.M.; Galaverni, M.; Fan, Z.; Marx, P.; Lorente-Galdos, B.; et al. Genome Sequencing Highlights the Dynamic Early History of Dogs. PLoS Genet. 2014, 10, e1004016. [Google Scholar] [CrossRef] [PubMed]
- García-García, G.; Baux, D.; Faugère, V.; Moclyn, M.; Koenig, M.; Claustres, M.; Roux, A.-F. Assessment of the Latest NGS Enrichment Capture Methods in Clinical Context. Sci. Rep. 2016, 6, 20948. [Google Scholar] [CrossRef] [PubMed]
- Mokry, M.; Feitsma, H.; Nijman, I.J.; de Bruijn, E.; van der Zaag, P.J.; Guryev, V.; Cuppen, E. Accurate SNP and Mutation Detection by Targeted Custom Microarray-Based Genomic Enrichment of Short-Fragment Sequencing Libraries. Nucleic Acids Res. 2010, 38, e116. [Google Scholar] [CrossRef]
- Gnirke, A.; Melnikov, A.; Maguire, J.; Rogov, P.; LeProust, E.M.; Brockman, W.; Fennell, T.; Giannoukos, G.; Fisher, S.; Russ, C.; et al. Solution Hybrid Selection with Ultra-Long Oligonucleotides for Massively Parallel Targeted Sequencing. Nat. Biotechnol. 2009, 27, 182–189. [Google Scholar] [CrossRef]
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.F.; Orlando, L. MapDamage2.0: Fast Approximate Bayesian Estimates of Ancient DNA Damage Parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef]
- Urios, V.; Donat-Torres, M.P.; Ramírez, C.; Monroy-Vilchis, O.; Rgribi-Idrissi, H. El Análisis Del Genoma Mitocondrial Del Cánido Estudiado En Marruecos Manifiesta Que No Es Ni Lobo (Canis lupus) Ni Chacal Euroasiático (Canis Aureus). AltoterO 2015, 3, 2–15. [Google Scholar]
- Björnerfeldt, S.; Webster, M.T.; Vilà, C. Relaxation of Selective Constraint on Dog Mitochondrial DNA Following Domestication. Genome Res. 2006, 16, 990–994. [Google Scholar] [CrossRef]
- Pang, J.-F.; Kluetsch, C.; Zou, X.-J.; Zhang, A.; Luo, L.-Y.; Angleby, H.; Ardalan, A.; Ekström, C.; Sköllermo, A.; Lundeberg, J.; et al. MtDNA Data Indicate a Single Origin for Dogs South of Yangtze River, Less than 16,300 Years Ago, from Numerous Wolves. Mol. Biol. Evol. 2009, 26, 2849–2864. [Google Scholar] [CrossRef]
- Thalmann, O.; Shapiro, B.; Cui, P.; Schuenemann, V.J.; Sawyer, S.K.; Greenfield, D.L.; Germonpré, M.B.; Sablin, M.V.; López-Giráldez, F.; Domingo-Roura, X.; et al. Complete Mitochondrial Genomes of Ancient Canids Suggest a European Origin of Domestic Dogs. Science 2013, 342, 871–874. [Google Scholar] [CrossRef]
- Arnason, U.; Gullberg, A.; Janke, A.; Kullberg, M. Mitogenomic Analyses of Caniform Relationships. Mol. Phylogenetics Evol. 2007, 45, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Campana, M.G.; Parker, L.D.; Hawkins, M.T.R.; Young, H.S.; Helgen, K.M.; Szykman Gunther, M.; Woodroffe, R.; Maldonado, J.E.; Fleischer, R.C. Genome Sequence, Population History, and Pelage Genetics of the Endangered African Wild Dog (Lycaon pictus). BMC Genom. 2016, 17, 1013. [Google Scholar] [CrossRef] [PubMed]
- Kazuno, A.-A.; Munakata, K.; Mori, K.; Tanaka, M.; Nanko, S.; Kunugi, H.; Umekage, T.; Tochigi, M.; Kohda, K.; Sasaki, T.; et al. Mitochondrial DNA Sequence Analysis of Patients with ‘Atypical Psychosis’. Psychiatry Clin. Neurosci. 2005, 59, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Hofman, C.A.; Rick, T.C.; Hawkins, M.T.R.; Funk, W.C.; Ralls, K.; Boser, C.L.; Collins, P.W.; Coonan, T.; King, J.L.; Morrison, S.A.; et al. Mitochondrial Genomes Suggest Rapid Evolution of Dwarf California Channel Islands Foxes (Urocyon littoralis). PLoS ONE 2015, 10, e0118240. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian Inference of Phylogenetic Trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.-X.; Hou, X.-D.; Barlow, A.; Preick, M.; Taron, U.H.; Alberti, F.; Basler, N.; Deng, T.; Lai, X.-L.; Hofreiter, M.; et al. Molecular Identification of Late and Terminal Pleistocene Equus ovodovi from Northeastern China. PLoS ONE 2019, 14, e0216883. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-Joining Networks for Inferring Intraspecific Phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.W.; Bryant, D. Popart: Full-Feature Software for Haplotype Network Construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.F.; Green, R.E.; Kelso, J.; Prüfer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of Damage in Genomic DNA Sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef]
- Meyer, M.; Kircher, M.; Gansauge, M.-T.; Li, H.; Racimo, F.; Mallick, S.; Schraiber, J.G.; Jay, F.; Prüfer, K.; de Filippo, C.; et al. A High-Coverage Genome Sequence from an Archaic Denisovan Individual. Science 2012, 338, 222–226. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Paijmans, J.L.A.; Barnett, R.; Gilbert, M.T.P.; Zepeda-Mendoza, M.L.; Reumer, J.W.F.; de Vos, J.; Zazula, G.; Nagel, D.; Baryshnikov, G.F.; Leonard, J.A.; et al. Evolutionary History of Saber-Toothed Cats Based on Ancient Mitogenomics. Curr. Biol. 2017, 27, 3330–3336.e5. [Google Scholar] [CrossRef]
- Krause, J.; Fu, Q.; Good, J.M.; Viola, B.; Shunkov, M.V.; Derevianko, A.P.; Pääbo, S. The Complete Mitochondrial DNA Genome of an Unknown Hominin from Southern Siberia. Nature 2010, 464, 894–897. [Google Scholar] [CrossRef]
- Grealy, A.C.; McDowell, M.C.; Scofield, P.; Murray, D.C.; Fusco, D.A.; Haile, J.; Prideaux, G.J.; Bunce, M. A Critical Evaluation of How Ancient DNA Bulk Bone Metabarcoding Complements Traditional Morphological Analysis of Fossil Assemblages. Quat. Sci. Rev. 2015, 128, 37–47. [Google Scholar] [CrossRef]
- Murray, D.C.; Haile, J.; Dortch, J.; White, N.E.; Haouchar, D.; Bellgard, M.I.; Allcock, R.J.; Prideaux, G.J.; Bunce, M. Scrapheap Challenge: A Novel Bulk-Bone Metabarcoding Method to Investigate Ancient DNA in Faunal Assemblages. Sci. Rep. 2013, 3, 3371. [Google Scholar] [CrossRef] [PubMed]
- Tautz, D.; Arctander, P.; Minelli, A.; Thomas, R.H.; Vogler, A.P. A Plea for DNA Taxonomy. Trends Ecol. Evol. 2003, 18, 70–74. [Google Scholar] [CrossRef]
- Basler, N.; Xenikoudakis, G.; Westbury, M.V.; Song, L.; Sheng, G.; Barlow, A. Reduction of the Contaminant Fraction of DNA Obtained from an Ancient Giant Panda Bone. BMC Res. Notes 2017, 10, 754. [Google Scholar] [CrossRef] [PubMed]
- Taron, U.H.; Lell, M.; Barlow, A.; Paijmans, J.L.A. Testing of Alignment Parameters for Ancient Samples: Evaluating and Optimizing Mapping Parameters for Ancient Samples Using the TAPAS Tool. Genes 2018, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Paijmans, J.L.A.; Fickel, J.; Courtiol, A.; Hofreiter, M.; Förster, D.W. Impact of Enrichment Conditions on Cross-Species Capture of Fresh and Degraded DNA. Mol. Ecol. Resour. 2016, 16, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Paijmans, J.L.A.; Barlow, A.; Henneberger, K.; Fickel, J.; Hofreiter, M.; Foerster, D.W.G. Ancestral Mitogenome Capture of the Southeast Asian Banded Linsang. PLoS ONE 2020, 15, e0234385. [Google Scholar] [CrossRef]
- Martinez, P.A.; Zurano, J.P.; Molina, W.F.; Bidau, C.J. Applications and Implications of Phylogeography for Canid Conservation. Mastozoología Neotropical 2013, 20, 61–74. [Google Scholar]
- Hailer, F.; Leonard, J.A. Hybridization among Three Native North American Canis Species in a Region of Natural Sympatry. PLoS ONE 2008, 3, e3333. [Google Scholar] [CrossRef]
- Wang, G.-D.; Zhang, M.; Wang, X.; Yang, M.A.; Cao, P.; Liu, F.; Lu, H.; Feng, X.; Skoglund, P.; Wang, L.; et al. Genomic Approaches Reveal an Endemic Subpopulation of Gray Wolves in Southern China. iScience 2019, 20, 110–118. [Google Scholar] [CrossRef]
- Ehrlinger, S.; Zenger, K. Ein Cuon-Fund Aus Der Zoolithenhöhle—Morphologische Und Biostatistische Studien Unter Verwendung Einer ACCESS-Datenbank. In Quartär: Internationales Jahrbuch zur Eiszeitalter- und Steinzeitforschung/International Yearbook for Ice Age and Stone Age Research; Verlag Marie Leidorf GmbH: Rahden, Germany, 1999; Volume 49–50, pp. 55–86. [Google Scholar]
- Nehring, A. Über Cuon alpinus fossilis nebst Bemerkungen über einige andere fossile Caniden. In Neues Jahrbuch für Mineralogie, Geologie und Palaeontologie; E. Schweizerbart’sche Verlagshandlung: Stuttgart, Germany, 1890; Volume 2, pp. 34–52. [Google Scholar]
- Buchalczyk, T.; Dynowski, J.; Szteyn, S. Variations in Number of Teeth and Asymmetry of the Skull in the Wolf. Acta Theriol. 1981, 26, 23–30. [Google Scholar] [CrossRef]
- Andersone, Z.; Ozolins, J. Craniometrical Characteristics and Dental Anomalies in Wolves Canis lupus from Latvia. Acta Theriol. 2000, 45, 549–558. [Google Scholar] [CrossRef]
- Mallye, J.-B.; Costamagno, S.; Boudadi-Maligne, M.; Prucca, A.; Lauroulandie, C. Dhole (Cuon alpinus) as a bone accumulator and new taphonomy agent? The case of Noisetier cave (French Pyrenees). J. Taphon. 2012, 10, 317–347. [Google Scholar]
- Baryshnikov, G.F. Late Pleistocene Canidae Remains from Geographical Society Cave in the Russian Far East. Russ. J. Theriol. 2015, 14, 65–83. [Google Scholar] [CrossRef]
- Liu, W.; Wu, X.; Pei, S.; Wu, X.; Norton, C.J. Huanglong Cave: A Late Pleistocene Human Fossil Site in Hubei Province, China. Quat. Int. 2010, 211, 29–41. [Google Scholar] [CrossRef]
- Qinqi, X. Some remarks on Chenjiawo fauna-On the First Appearance of Peking Man Fauna. Vertebr. Pal Asiatica 1996, 34, 41–57. [Google Scholar]
- Weiwen, H.; Xinqiang, S.; Yamei, H.; Miller-Antonio, S.; Schepartz, L.A. Excavations at Panxian Dadong, Guizhou Province, Southern China. Curr. Anthropol. 1995, 36, 844–846. [Google Scholar] [CrossRef]
- Suraprasit, K.; Jaeger, J.-J.; Chaimanee, Y.; Chavasseau, O.; Yamee, C.; Tian, P.; Panha, S. The Middle Pleistocene Vertebrate Fauna from Khok Sung (Nakhon Ratchasima, Thailand): Biochronological and Paleobiogeographical Implications. Zookeys 2016, 1–157. [Google Scholar] [CrossRef]
- Long, V.T.; de Vos, J.; Ciochon, R.L. The Fossil Mammal Fauna of the Lang Trang Caves, Vietnam, Compared with Southeast Asian Fossil and Recent Mammal Faunas: The Geographic Implications. Bull. Indo-Pac. Prehistory Assoc. 1996, 14, 101–109. [Google Scholar]
- Hofreiter, M. Long DNA Sequences and Large Data Sets: Investigating the Quaternary via Ancient DNA. Quat. Sci. Rev. 2008, 27, 2586–2592. [Google Scholar] [CrossRef]
- Hofreiter, M.; Barnes, I. Diversity Lost: Are All Holarctic Large Mammal Species Just Relict Populations? BMC Biol. 2010, 8, 46. [Google Scholar] [CrossRef]
- De Bruyn, M.; Hoelzel, A.R.; Carvalho, G.R.; Hofreiter, M. Faunal Histories from Holocene Ancient DNA. Trends Ecol. Evol. 2011, 26, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.A.; Vilà, C.; Fox-Dobbs, K.; Koch, P.L.; Wayne, R.K.; Van Valkenburgh, B. Megafaunal Extinctions and the Disappearance of a Specialized Wolf Ecomorph. Curr. Biol. 2007, 17, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Pilot, M.; Branicki, W.; Jędrzejewski, W.; Goszczyński, J.; Jędrzejewska, B.; Dykyy, I.; Shkvyrya, M.; Tsingarska, E. Phylogeographic History of Grey Wolves in Europe. BMC Evol. Biol. 2010, 10, 104. [Google Scholar] [CrossRef] [PubMed]
- Sheng, G.-L.; Barlow, A.; Cooper, A.; Hou, X.-D.; Ji, X.-P.; Jablonski, N.G.; Zhong, B.-J.; Liu, H.; Flynn, L.J.; Yuan, J.-X.; et al. Ancient DNA from Giant Panda (Ailuropoda melanoleuca) of South-Western China Reveals Genetic Diversity Loss during the Holocene. Genes 2018, 9, 198. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, J.L.; Turney, C.; Barnett, R.; Martin, F.; Bray, S.C.; Vilstrup, J.T.; Orlando, L.; Salas-Gismondi, R.; Loponte, D.; Medina, M.; et al. Synergistic Roles of Climate Warming and Human Occupation in Patagonian Megafaunal Extinctions during the Last Deglaciation. Sci. Adv. 2016, 2, e1501682. [Google Scholar] [CrossRef]
- Karanth, K.U.; Sunquist, M.E. Prey Selection by Tiger, Leopard and Dhole in Tropical Forests. J. Anim. Ecol. 1995, 64, 439–450. [Google Scholar] [CrossRef]
- Kamler, J.F.; Johnson, A.; Vongkhamheng, C.; Bousa, A. The Diet, Prey Selection, and Activity of Dholes (Cuon alpinus) in Northern Laos. J. Mammal. 2012, 93, 627–633. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Element | Location | Approx. Age | Reference |
|---|---|---|---|---|
| Y-37 | mandible | Jáchymka cave, Czech Republic | ca. 35–45 ka (co-occurring remains have been dated, Marciszak, unpublished data) | [25] |
| Y-38 | skull | Jáchymka cave, Czech Republic | ca. 35–45 ka (co-occurring remains have been dated, Marciszak, unpublished data) | [25] |
| Y-39 | mandible | Bacho Kiro Cave, Bulgaria | ca. 39–45 ka (multiple samples from the same layer have been dated, e.g., [28]) | [26] |
| C3-6-2 | metapodial | Peștera Seacă din Ogașul Stoienilor, Romania | ca. 25 ka (a mandible found close by was dated) | [27] |
| C3-6-3 | metapodial | Peștera Seacă din Ogașul Stoienilor, Romania | ca. 25 ka (a mandible found close by was dated) | [27] |
| C3-6-4 | metapodial | Peștera Seacă din Ogașul Stoienilor, Romania | ca. 25 ka (a mandible found close by was dated) | [27] |
| modern dhole | blood (provided as DNA extract) | Allwetterzoo Münster | Recent | [21] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taron, U.H.; Paijmans, J.L.A.; Barlow, A.; Preick, M.; Iyengar, A.; Drăgușin, V.; Vasile, Ș.; Marciszak, A.; Roblíčková, M.; Hofreiter, M. Ancient DNA from the Asiatic Wild Dog (Cuon alpinus) from Europe. Genes 2021, 12, 144. https://doi.org/10.3390/genes12020144
Taron UH, Paijmans JLA, Barlow A, Preick M, Iyengar A, Drăgușin V, Vasile Ș, Marciszak A, Roblíčková M, Hofreiter M. Ancient DNA from the Asiatic Wild Dog (Cuon alpinus) from Europe. Genes. 2021; 12(2):144. https://doi.org/10.3390/genes12020144
Chicago/Turabian StyleTaron, Ulrike H., Johanna L. A. Paijmans, Axel Barlow, Michaela Preick, Arati Iyengar, Virgil Drăgușin, Ștefan Vasile, Adrian Marciszak, Martina Roblíčková, and Michael Hofreiter. 2021. "Ancient DNA from the Asiatic Wild Dog (Cuon alpinus) from Europe" Genes 12, no. 2: 144. https://doi.org/10.3390/genes12020144
APA StyleTaron, U. H., Paijmans, J. L. A., Barlow, A., Preick, M., Iyengar, A., Drăgușin, V., Vasile, Ș., Marciszak, A., Roblíčková, M., & Hofreiter, M. (2021). Ancient DNA from the Asiatic Wild Dog (Cuon alpinus) from Europe. Genes, 12(2), 144. https://doi.org/10.3390/genes12020144