
The Genetics of Inherited Cholestatic Disorders in Neonates and Infants: Evolving Challenges

 ,
,  and
and
Abstract
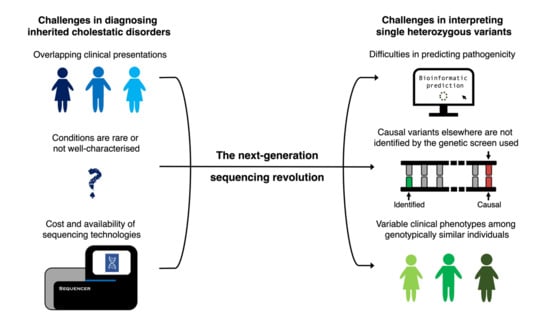
:
1. Introduction
2. Normal Bile Secretion and Flow
3. Genetic Causes of Cholestasis in Neonates and Infants
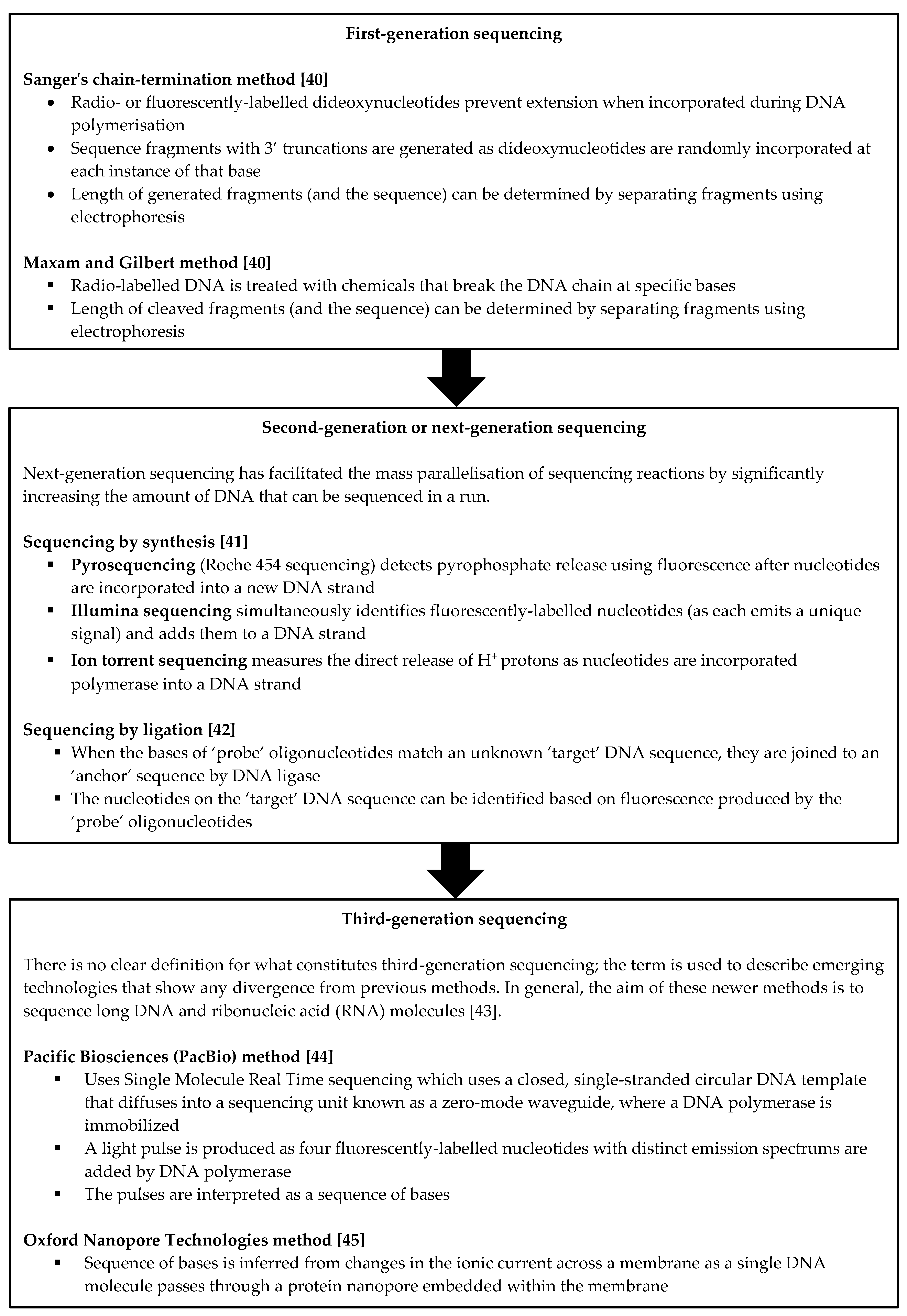
4. The Role of Next-Generation Sequencing in the Work-Up of Cholestasis
5. New Diagnostic Challenges in the Era of Sequencing
5.1. Frequency of Heterozygous Pathogenic Variants in Children with Undiagnosed Liver Disease
5.1.1. ATP8B1, ABCB11 and ABCB4 Mutations
5.1.2. NPC1 and NPC2 Mutations
5.1.3. SLC25A13 Mutations
5.2. Difficulties in Predicting Pathogenicity
5.3. Mutations Elsewhere That Have Not Been Identified
5.4. Variable Clinical Phenotypes
5.5. Practical Considerations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A1AT | α1-antitrypsin |
| ABCB4 | ATP binding cassette subfamily B member 4 |
| ABCB11 | ATP binding cassette subfamily B member 11 |
| AGVGD | Align GVGD |
| ALF | acute liver failure |
| ATP8B1 | ATPase phospholipid transporting 8B1 |
| BA | biliary atresia |
| BRIC | benign recurrent intrahepatic cholestasis |
| BSEP | bile salt export pump |
| DIC | drug-induced cholestasis |
| DNA | deoxyribonucleic acid |
| GRACILE | Growth Retardation, Aminoaciduria, Cholestasis, Iron overload, Lactic acidosis, and Early death |
| GS | Genesplicer |
| HSF | Human Splicing Finder |
| IBAT | ileal bile acid transporter |
| ICP | intrahepatic cholestasis of pregnancy |
| KIF12 | kinesin family member 12 |
| LT | liver transplant |
| LPAC | low phospholipid-associated cholelithiasis |
| LSR | lipolysis stimulated lipoprotein receptor |
| MDR3 | multidrug resistance protein 3 |
| MES | MaxEntScan |
| MS | microarray resequencing |
| MYO5B | myosin VB |
| NGS | next-generation sequencing |
| NICCD | neonatal intrahepatic cholestasis due to citrin deficiency |
| NNS | NNSplice |
| NPC | Niemann–Pick disease type C |
| NPC-1 | Niemann–Pick disease type C1 |
| NPC1 | NPC intracellular cholesterol transporter 1 |
| NPC2 | NPC intracellular cholesterol transporter 2 |
| PEBD | partial extrahepatic biliary diversion |
| PFIC | progressive familial intrahepatic cholestasis |
| PP | PolyPhen-2 |
| RAB | Ras-related in brain |
| RNA | ribonucleic acid |
| SIFT | Sorting Intolerant from Tolerant |
| SLC25A13 | solute carrier family 25 member 13 |
| SSF | SpliceSiteFinder-like |
| TNC | transient neonatal cholestasis |
References
- Maddirevula, S.; Alhebbi, H.; Alqahtani, A.; Algoufi, T.; AlSaif, H.S.; Ibrahim, N.; Abdulwahab, F.; Barr, M.; Alzaidan, H.; Almehaideb, A.; et al. Identification of novel loci for pediatric cholestatic liver disease defined by KIF12, PPM1F, USP53, LSR, and WDR83OS pathogenic variants. Genet. Med. 2018, 21, 1164–1172. [Google Scholar] [CrossRef]
- Gonzales, E.; Taylor, S.A.; Davit-Spraul, A.; Thébaut, A.; Thomassin, N.; Guettier, C.; Whitington, P.F.; Jacquemin, E. MYO5B mutations cause cholestasis with normal serum γ-glutamyl transferase activity in children without microvillous inclusion disease. Hepatology 2016, 65, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.L. Bile Formation and Secretion. Compr. Physiol. 2013, 3, 1035–1078. [Google Scholar]
- Boyer, J.L.; Soroka, C.J. Bile formation and secretion: An update. J. Hepatol. 2021, 75, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.; Jesú Prieto, J.F.M.S. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J. Gastroenterol. 2006, 12, 3496–3511. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Garruti, G.; Baccetto, R.L.; Molina-Molina, E.; Bonfrate, L.; Portincasa, P.; Wang, D.Q. Bile Acid Physiology. Ann. Hepatol. 2017, 16 (Suppl. 1), S4–S14. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.-J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.G.; Sokol, R.J. Neonatal cholestasis: Emerging molecular diagnostics and potential novel therapeutics. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 346–360. [Google Scholar] [CrossRef]
- Goldberg, A.; Mack, C.L. Inherited Cholestatic Diseases in the Era of Personalized Medicine. Clin. Liver Dis. 2020, 15, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Feldman, A.G.; Sokol, R.J. Neonatal Cholestasis. Neoreviews 2013, 14, e63–e73. [Google Scholar] [CrossRef] [Green Version]
- Balistreri, W.F.; Bezerra, J.A. Whatever happened to “neonatal hepatitis”? Clin. Liver Dis. 2006, 10, 27–53. [Google Scholar] [CrossRef] [PubMed]
- National Organisation for Rare Diseases: Rare Diseases Database. Neonatal Cholestasis. 2020. Available online: https://rarediseases.org/rare-diseases/idiopathic-neonatal-hepatitis/ (accessed on 14 November 2021).
- Treepongkaruna, S.; Jitraruch, S.; Kodcharin, P.; Charoenpipop, D.; Suwannarat, P.; Pienvichit, P.; Kobayashi, K.; Wattanasirichaigoon, D. Neonatal intrahepatic cholestasis caused by citrin deficiency: Prevalence and SLC25A13 mutations among Thai infants. BMC Gastroenterol. 2012, 12, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.B.; Kobayashi, K.; Ushikai, M.; Tabata, A.; Iijima, M.; Li, M.X.; Lei, L.; Kawabe, K.; Taura, S.; Yang, Y.; et al. Frequency and distribution in East Asia of 12 mutations identified in the SLC25A13 gene of Japanese patients with citrin deficiency. J. Hum. Genet. 2005, 50, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Abuduxikuer, K.; Chen, R.; Wang, Z.-L.; Wang, J. Risk factors associated with mortality in neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) and clinical implications. BMC Pediatr. 2019, 19, 18. [Google Scholar] [CrossRef] [Green Version]
- Millat, G.; Marçais, C.; Rafi, M.A.; Yamamoto, T.; Morris, J.A.; Pentchev, P.G.; Ohno, K.; Wenger, D.A.; Vanier, M.T. Niemann-Pick C1 Disease: The I1061T Substitution Is a Frequent Mutant Allele in Patients of Western European Descent and Correlates with a Classic Juvenile Phenotype. Am. J. Hum. Genet. 1999, 65, 1321–1329. [Google Scholar] [CrossRef] [Green Version]
- Alagille, D.; Odièvre, M.; Gautier, M.; Dommergues, J. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. J. Pediatr. 1975, 86, 63–71. [Google Scholar] [CrossRef]
- Nemes, K.; Åberg, F.; Gylling, H.; Isoniemi, H. Cholesterol metabolism in cholestatic liver disease and liver transplantation: From molecular mechanisms to clinical implications. World J. Hepatol. 2016, 8, 924–932. [Google Scholar] [CrossRef]
- Sundaram, S.S.; Bove, K.E.; Lovell, M.A.; Sokol, R.J. Mechanisms of Disease: Inborn errors of bile acid synthesis. Nat. Clin. Pr. Gastroenterol. Hepatol. 2008, 5, 456–468. [Google Scholar] [CrossRef] [Green Version]
- Gissen, P.; Johnson, C.A.; Morgan, N.V.; Stapelbroek, J.M.; Forshew, T.; Cooper, W.N.; Maher, E.R. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat. Genet. 2004, 36, 400–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlton, V.E.; Knisely, A.; Freimer, N.B. Mapping of a locus for progressive familial intrahepatic cholestasis (Byler disease) to 18q21-q22, the benign recurrent intrahepatic cholestasis region. Hum. Mol. Genet. 1995, 4, 1049–1053. [Google Scholar] [CrossRef]
- Bull, L.N.; Van Eijk, M.J.T.; Pawlikowska, L.; Deyoung, J.A.; Juijn, J.A.; Liao, M.; Klomp, L.W.J.; Lomri, N.; Berger, R.; Scharschmidt, B.R.; et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat. Genet. 1998, 18, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Strautnieks, S.S.; Bull, L.N.; Knisely, A.S.; Kocoshis, S.A.; Dahl, N.; Arnell, H.; Sokal, E.; Dahan, K.; Childs, S.; Ling, V.; et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat. Genet. 1998, 20, 233–238. [Google Scholar] [CrossRef]
- Deleuze, J.F.; Jacquemin, E.; Dubuisson, C.; Cresteil, D.; Dumont, M.; Erlinger, S.; Hadchouel, M. Defect of multidrug-resistance 3 gene expression in a subtype of progressive familial intrahepatic cholestasis. Hepatology 1996, 23, 904–908. [Google Scholar] [CrossRef]
- Memon, N.; Weinberger, B.I.; Hegyi, T.; Aleksunes, L. Inherited disorders of bilirubin clearance. Pediatr. Res. 2015, 79, 378–386. [Google Scholar] [CrossRef] [Green Version]
- van de Steeg, E.; Stránecký, V.; Hartmannová, H.; Nosková, L.; Hřebíček, M.; Wagenaar, E.; Schinkel, A.H. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J. Clin. Investig. 2012, 122, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Paulusma, C.; Kool, M.; Bosma, P.J.; Scheffer, G.L.; Ter Borg, F.; Scheper, R.J.; Tytgat, G.N.; Borst, P.; Baas, F.; Elferink, R.P.O. A mutation in the human canalicular multispecific organic anion transporter gene causes the Dubin-Johnson syndrome. Hepatology 1997, 25, 1539–1542. [Google Scholar] [CrossRef] [Green Version]
- Wada, M.; Toh, S.; Taniguchi, K.; Nakamura, T.; Uchiumi, T.; Kohno, K.; Yoshida, I.; Kimura, A.; Sakisaka, S.; Adachi, Y.; et al. Mutations in the canilicular multispecific organic anion transporter (cMOAT) gene, a novel ABC transporter, in patients with hyperbilirubinemia II/Dubin-Johnson syndrome. Hum. Mol. Genet. 1998, 7, 203–207. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.; Horslen, S. Metabolic liver disease in children. Liver Transpl. 2008, 14, 713–733. [Google Scholar] [CrossRef]
- Gunaydin, M.; Cil, A.T.B. Cholestasis in the baby and infant. EMJ 2019, 4, 73–82. [Google Scholar]
- Clayton, P.T. Inborn errors presenting with liver dysfunction. Semin. Neonatol. 2002, 7, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Sokol, R.J. Liver Disease in Mitochondrial Disorders. Semin. Liver Dis. 2007, 27, 259–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visapää, I.; Fellman, V.; Vesa, J.; Dasvarma, A.; Hutton, J.L.; Kumar, V.; Payne, G.S.; Makarow, M.; Van Coster, R.; Taylor, R.W.; et al. GRACILE Syndrome, a Lethal Metabolic Disorder with Iron Overload, Is Caused by a Point Mutation in BCS1L. Am. J. Hum. Genet. 2002, 71, 863–876. [Google Scholar] [CrossRef] [Green Version]
- Pandita, A.; Gupta, V.; Gupta, G. Neonatal Cholestasis: A Pandora’s Box. Clin. Med. Insights Pediatr. 2018, 12, 1179556518805412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fawaz, R.; Baumann, U.; Ekong, U.; Fischler, B.; Hadzic, N.; Mack, C.L.; McLin, V.A.; Molleston, J.P.; Neimark, E.; Ng, V.L.; et al. Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 154–168. [Google Scholar]
- Nicastro, E.; Di Giorgio, A.; Marchetti, D.; Barboni, C.; Cereda, A.; Iascone, M.; D’Antiga, L. Diagnostic Yield of an Algorithm for Neonatal and Infantile Cholestasis Integrating Next-Generation Sequencing. J. Pediatr. 2019, 211, 54–62.e4. [Google Scholar] [CrossRef]
- Shimura, M.; Kuranobu, N.; Ogawa-Tominaga, M.; Akiyama, N.; Sugiyama, Y.; Ebihara, T.; Fushimi, T.; Ichimoto, K.; Matsunaga, A.; Tsuruoka, T.; et al. Clinical and molecular basis of hepatocerebral mitochondrial DNA depletion syndrome in Japan: Evaluation of outcomes after liver transplantation. Orphanet J. Rare Dis. 2020, 15, 169. [Google Scholar] [CrossRef]
- Grabhorn, E.; Tsiakas, K.; Herden, U.; Fischer, L.; Freisinger, P.; Marquardt, T.; Ganschow, R.; Briem-Richter, A.; Santer, R. Long-term outcomes after liver transplantation for deoxyguanosine kinase deficiency: A single-center experience and a review of the literature. Liver Transplant. 2014, 20, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Modin, L.; Ng, V.; Gissen, P.; Raiman, J.; Pfister, E.D.; Das, A.; Santer, R.; Faghfoury, H.; Santra, S.; Baumann, U. A Case Series on Genotype and Outcome of Liver Transplantation in Children with Niemann-Pick Disease Type C. Children 2021, 8, 819. [Google Scholar] [CrossRef]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2015, 107, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Ho, A.; Murphy, M.; Wilson, S.; Atlas, S.R.; Edwards, J.S. Sequencing by ligation variation with endonuclease V digestion and deoxyinosine-containing query oligonucleotides. BMC Genom. 2011, 12, 598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y. Advent of a new sequencing era: Long-read and on-site sequencing. J. Hum. Genet. 2020, 65, 1. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinf. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.-Y.; Wang, X.-H.; Lu, Y.; Zhu, Q.-R.; Wang, J.-S. Association of variants of ABCB11 with transient neonatal cholestasis. Pediatr. Int. 2013, 55, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, E.; Malan, V.; Rio, M.; Davit-Spraul, A.; Cohen, J.; Landrieu, P.; Bernard, O. Heterozygous FIC1 Deficiency: A New Genetic Predisposition to Transient Neonatal Cholestasis. J. Pediatr. Gastroenterol. Nutr. 2010, 50, 447–449. [Google Scholar] [CrossRef]
- Hijikata, A.; Tsuji, T.; Shionyu, M.; Shirai, T. Decoding disease-causing mechanisms of missense mutations from supramolecular structures. Sci. Rep. 2017, 7, 8541. [Google Scholar] [CrossRef] [PubMed]
- McKay, K.E.; Bruce, C.K.; Hartley, J.L.; Knisely, A.S.; Baumann, U.; Bockisch, S.S.; Gissen, P. Mutation detection in cholestatic patients using microarray resequencing of ATP8B1 and ABCB11. F1000Research 2013, 2, 32. [Google Scholar] [CrossRef]
- Bounford, K. Investigations into the Genetic Causes of Liver Disease Using Molecular Genetic Technologies. University of Birmingham. 2016. Available online: http://etheses.bham.ac.uk//id/eprint/7101/ (accessed on 14 November 2021).
- Tamura, H.; Takahashi, T.; Ban, N.; Torisu, H.; Ninomiya, H.; Takada, G.; Inagaki, N. Niemann–Pick type C disease: Novel NPC1 mutations and characterization of the concomitant acid sphingomyelinase deficiency. Mol. Genet. Metab. 2006, 87, 113–121. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nanba, E.; Ninomiya, H.; Higaki, K.; Taniguchi, M.; Zhang, H.; Ohno, K. NPC1 gene mutations in Japanese patients with Niemann-Pick disease type C. Hum. Genet. 1999, 105, 10–16. [Google Scholar]
- Ganesh, R.; Suresh, N.; Sathiyasekeran, M.; Venkatakrishnan, L. Benign Recurrent Intrahepatic Cholestasis—Unravelleing the Paradox. Indian Pediatr. 2021, 58, 486–487. [Google Scholar] [CrossRef] [PubMed]
- Klomp, L.W.J.; Vargas, J.C.; Van Mil, S.W.C.; Pawlikowska, L.; Strautnieks, S.S.; Van Eijk, M.J.T.; Juijn, J.A.; Pabón-Peña, C.; Smith, L.B.; DeYoung, J.A.; et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology 2004, 40, 27–38. [Google Scholar] [CrossRef]
- Strautnieks, S.S.; Byrne, J.A.; Pawlikowska, L.; Cebecauerová, D.; Rayner, A.; Dutton, L.; Thompson, R.J. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology 2008, 134, 1203–1214. [Google Scholar] [CrossRef] [Green Version]
- Knisely, A.S.; Strautnieks, S.S.; Meier, Y.; Stieger, B.; Byrne, J.A.; Portmann, B.C.; Bull, L.N.; Pawlikowska, L.; Bilezikçi, B.; Özçay, F. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology. 2006, 44, 478–486. [Google Scholar] [CrossRef]
- Kubitz, R.; Dröge, C.; Stindt, J.; Weissenberger, K.; Häussinger, D. The bile salt export pump (BSEP) in health and disease. Clin. Res. Hepatol. Gastroenterol. 2012, 36, 536–553. [Google Scholar] [CrossRef]
- Byrne, J.A.; Strautnieks, S.S.; Ihrke, G.; Pagani, F.; Knisely, A.; Linton, K.J.; Mieli-Vergani, G.; Thompson, R.J. Missense mutations and single nucleotide polymorphisms in ABCB11 impair bile salt export pump processing and function or disrupt pre-messenger RNA splicing. Hepatology 2008, 49, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, E.; Grosse, B.; Cassio, D.; Davit-Spraul, A.; Fabre, M.; Jacquemin, E. Successful mutation-specific chaperone therapy with 4-phenylbutyrate in a child with progressive familial intrahepatic cholestasis type 2. J. Hepatol. 2012, 57, 695–698. [Google Scholar] [CrossRef]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-Pick C1 Disease Gene: Homology to Mediators of Cholesterol Homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Nobili, V.; Di Giandomenico, S.; Francalanci, P.; Callea, F.; Marcellini, M.; Santorelli, F.M. A new ABCB11 mutation in two Italian children with familial intrahepatic cholestasis. J. Gastroenterol. 2006, 41, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Davit-Spraul, A.; Gonzales, E.; Baussan, C.; Jacquemin, E. The Spectrum of Liver Diseases Related to ABCB4 Gene Mutations: Pathophysiology and Clinical Aspects. Semin. Liver Dis. 2010, 30, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.; Balding, D.; Klünemann, H.H.; Linden, D.; Ory, D.S.; Pineda, M.; Priller, J.; Sedel, F.; Muller, A.; Chadha-Boreham, H.; et al. Genetic screening for Niemann-Pick disease type C in adults with neurological and psychiatric symptoms: Findings from the ZOOM study. Hum. Mol. Genet. 2013, 22, 4349–4356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyre, D.; Delplanque, J.; Chèvre, J.-C.; Lecoeur, C.; Lobbens, S.; Gallina, S.; Durand, E.; Vatin, V.; Degraeve, F.; Proença, C.; et al. Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat. Genet. 2009, 41, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Lamri, A.; Pigeyre, M.; Garver, W.S.; Meyre, D. The Extending Spectrum of NPC1-Related Human Disorders: From Niemann–Pick C1 Disease to Obesity. Endocr. Rev. 2018, 39, 192–220. [Google Scholar] [CrossRef] [PubMed]
- Baghdasaryan, A.; Ofner-Ziegenfuß, L.; Lackner, C.; Fickert, P.; Resch, B.; Morris, N.M.; Deutschmann, A. Histological demonstration of BSEP/ABCB11 inhibition in transient neonatal cholestasis: A case report. BMC Pediatr. 2020, 20, 340. [Google Scholar] [CrossRef]
- Zhang, Z.-H.; Lin, W.-X.; Deng, M.; Zhao, S.-T.; Zeng, H.-S.; Chen, F.-P.; Song, Y.-Z. Clinical, Molecular and Functional Investigation on an Infant with Neonatal Intrahepatic Cholestasis Caused by Citrin Deficiency (NICCD). PLoS ONE 2014, 9, e89267. [Google Scholar]
- Park, H.J.; Kim, T.H.; Kim, S.W.; Noh, S.H.; Cho, K.J.; Choi, C.; Kwon, E.Y.; Choi, Y.J.; Gee, H.Y.; Choi, J.H. Functional characterization of ABCB4 mutations found in progressive familial intrahepatic cholestasis type 3. Sci. Rep. 2016, 6, 26872. [Google Scholar] [CrossRef] [Green Version]
- Ellard, S.; Colclough, K.; Patel, K.A.; Hattersley, A.T. Prediction algorithms: Pitfalls in interpreting genetic variants of autosomal dominant monogenic diabetes. J. Clin. Investig. 2019, 130, 14–16. [Google Scholar] [CrossRef] [Green Version]
- Strnad, P.; Buch, S.; Hamesch, K.; Fischer, J.; Rosendahl, J.; Schmelz, R.; Brueckner, S.; Brosch, M.; Heimes, C.V.; Woditsch, V.; et al. Heterozygous carriage of the alpha1-antitrypsin Pi*Z variant increases the risk to develop liver cirrhosis. Gut 2018, 68, 1099–1107. [Google Scholar] [CrossRef]
- Bartlett, J.R.; Friedman, K.J.; Ling, S.C.; Pace, R.G.; Bell, S.C.; Bourke, B.; Gene Modifier Study Group. Genetic modifiers of liver disease in cystic fibrosis. JAMA 2009, 302, 1076–1083. [Google Scholar] [CrossRef]
- Przybylla, S.; Stindt, J.; Kleinschrodt, D.; Schulte Am Esch, J.; Häussinger, D.; Keitel, V.; Smits, S.H.; Schmitt, L. Analysis of the Bile Salt Export Pump (ABCB11) Interactome Employing Complementary Approaches. PLoS ONE 2016, 11, e0159778. [Google Scholar] [CrossRef]
- Ben Saad, A.; Vauthier, V.; Lapalus, M.; Mareux, E.; Bennana, E.; Durand-Schneider, A.M.; Falguières, T. RAB10 Interacts with ABCB4 and Regulates Its Intracellular Traffic. Int. J. Mol. Sci. 2021, 22, 7087. [Google Scholar] [CrossRef]
- Uehara, T.; Yamada, M.; Umetsu, S.; Nittono, H.; Suzuki, H.; Fujisawa, T.; Takenouchi, T.; Inui, A.; Kosaki, K. Biallelic Mutations in the LSR Gene Cause a Novel Type of Infantile Intrahepatic Cholestasis. J. Pediatr. 2020, 221, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Stalke, A.; Sgodda, M.; Cantz, T.; Skawran, B.; Lainka, E.; Hartleben, B.; Baumann, U.; Pfister, E. KIF12 Variants and Disturbed Hepatocyte Polarity in Children with a Phenotypic Spectrum of Cholestatic Liver Disease. Available online: https://doi.org/10.1016/j.jpeds.2021.09.019 (accessed on 20 September 2021).
- Cooper, D.N.; Krawczak, M.; Polychronakos, C.; Tyler-Smith, C.; Kehrer-Sawatzki, H. Where genotype is not predictive of phenotype: Towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Qual. Life Res. 2013, 132, 1077–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merryweather-Clarke, A.T.; Cadet, E.; Bomford, A.; Capron, D.; Viprakasit, V.; Miller, A.; McHugh, P.J.; Chapman, R.W.; Pointon, J.J.; Wimhurst, V.L.; et al. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum. Mol. Genet. 2003, 12, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Yang, Z.; Vatta, M.; Rampazzo, A.; Beffagna, G.; Pillichou, K.; Scherer, S.E.; Saffitz, J.; Kravitz, J.; Zareba, W.; et al. Compound and Digenic Heterozygosity Contributes to Arrhythmogenic Right Ventricular Cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Gordon, R.J.; Li, D.; Doyle, D.; Zaritsky, J.; Levine, M.A. Digenic Heterozygous Mutations in SLC34A3 and SLC34A1 Cause Dominant Hypophosphatemic Rickets with Hypercalciuria. J. Clin. Endocrinol. Metab. 2020, 105, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Lourembam, R.; Malik, R.; Bolia, R. Combined Mutations of Canalicular Transporter Proteins Causing Low Phospholipid-Associated Cholelithiasis and Transient Neonatal Cholestasis in an Infant. JPGN Rep. 2021, 2, e080. [Google Scholar] [CrossRef]
- Kagawa, T.; Watanabe, N.; Mochizuki, K.; Numari, A.; Ikeno, Y.; Itoh, J.; Tanaka, H.; Arias, I.M.; Mine, T. Phenotypic differences in PFIC2 and BRIC2 correlate with protein stability of mutant Bsep and impaired taurocholate secretion in MDCK II cells. Am. J. Physiol. Liver Physiol. 2008, 294, G58–G67. [Google Scholar] [CrossRef] [Green Version]
- Stättermayer, A.F.; Halilbasic, E.; Wrba, F.; Ferenci, P.; Trauner, M. Variants in ABCB4 (MDR3) across the spectrum of cholestatic liver diseases in adults. J. Hepatol. 2020, 73, 651–663. [Google Scholar] [CrossRef]
- Dixon, P.H.; Sambrotta, M.; Chambers, J.; Taylor-Harris, P.; Syngelaki, A.; Nicolaides, K.; Knisely, A.S.; Thompson, R.; Williamson, C. An expanded role for heterozygous mutations of ABCB4, ABCB11, ATP8B1, ABCC2 and TJP2 in intrahepatic cholestasis of pregnancy. Sci. Rep. 2017, 7, 11823. [Google Scholar] [CrossRef] [Green Version]
- Hertel, P.M.; Bull, L.N.; Thompson, R.J.; Goodrich, N.P.; Ye, W.; Magee, J.C.; Squires, R.H.; Bass, L.M.; Heubi, J.E.; Kim, G.E.; et al. Mutation Analysis and Disease Features at Presentation in a Multi-Center Cohort of Children with Monogenic Cholestasis. J. Pediatr. Gastroenterol. Nutr. 2021, 73, 169–177. [Google Scholar] [CrossRef]
- van Wessel, D.B.E.; Thompson, R.J.; Gonzales, E.; Jankowska, I.; Sokal, E.; Grammatikopoulos, T.; Verkade, H.J. Genotype correlates with the natural history of severe bile salt export pump deficiency. J. Hepatol. 2020, 73, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, R.J.; Jaecklin, T.; Vig, P.; Jankowska, I.; Kelly, D.; Mack, C.; Loomes, K.; Miethke, A.; Rajwal, S.; Setchell, K.; et al. Genotype and dose-dependent response to maralixibat in patients with bile salt export pump deficiency. J. Pediatric Gastroenterol. Nutrition 2020, 71, S435. [Google Scholar]
- Raffan, E.; Semple, R.K. Next generation sequencing—Implications for clinical practice. Br. Med. Bull. 2011, 99, 53–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinical Sequencing Exploratory Research (CSER) Consortium. Guide to Interpreting Genomic Reports: A Genomic Tool Kit. Available online: https://www.genome.gov/sites/default/files/media/files/2020-04/Guide_to_Interpreting_Genomic_Reports_Toolkit.pdf (accessed on 14 November 2021).
- Chapman, C.R.; Mehta, K.S.; Parent, B.; Caplan, A.L. Genetic discrimination: Emerging ethical challenges in the context of advancing technology. J. Law Biosci. 2019, 7, lsz016. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Mechanism | Examples (Not Exhaustive) | References |
|---|---|---|
| Biliary tract anomalies | Alagille syndrome | [17] |
| Defect in the synthesis of components of bile | Bile acid synthesis defects e.g., cerebrotendinous xanthomatosis (or sterol 27-hydroxylase deficiency) Cholesterol synthesis defects e.g., lathosterolosis | [18,19] |
| Defects in intracellular trafficking | Arthrogryposis-renal dysfunction-cholestasis syndrome MYO5B-associated intrahepatic cholestasis | [2,20] |
| Defects in the export of components of bile or in tight junction formation | Progressive familial intrahepatic cholestasis types 1—6 Rotor syndrome Dubin-Johnson syndrome | [21,22,23,24,25,26,27,28] |
| Metabolic disorders | Disorders of lipid metabolism e.g., Niemann-Pick disease type C, Wolman disease, Farber disease, Gaucher disease, cholesterol ester storage disease Disorders of carbohydrate metabolism e.g., galactosaemia, hereditary fructose intolerance, glycogen storage disease type IV Disorders of amino acid metabolism e.g., tyrosinaemia Peroxisomal disorders e.g., Zellweger syndrome, adrenoleukodystrophy Urea cycle disorders e.g., arginase deficiency, citrin deficiency Mitochondrial disorders e.g., mitochondrial DNA depletion syndrome, fatty acid oxidation defects, GRACILE (Growth Retardation, Aminoaciduria, Cholestasis, Iron overload, Lactic acidosis and Early death) syndrome | [29,30,31,32,33] |
| Miscellaneous disorders | α1-antitrypsin deficiency Cystic fibrosis Indian childhood cirrhosis Cholestasis of North American Indians Trisomy 13, 18, or 21 Turner syndrome | [10,29,30,31,32,34] |
| Patient | Mutation 1 | Novel or Reference | Prediction Tools | Mutation 2 | Novel or Reference | Prediction Tools | Diagnosis | Presenting Features | Features at Follow-Up |
|---|---|---|---|---|---|---|---|---|---|
| 20 | NPC1 c.2000C>T p.(S667L) | [51] | AGVGD C65 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS Cryptic acceptor HSF No changes | NPC1 c.3182T>C p.(I1061T) | [52] (rs80358259) | AGVGD C25 SIFT Deleterious PP Possibly damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | NPC | C; H | Not available |
| 40 | ATP8B1 c.1244A>G p.(Q415R) | Novel at time of study. Subsequently published in [53] | AGVGD C0 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | ATP8B1 c.1244A>G p.(Q415R) | Novel at time of study. Subsequently published in [53] | AGVGD C0 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | PFIC type 1 | C; H | Not available |
| 86 | ATP8B1 c.1367C>T p.(T456M) | [54] (rs121909104) | AGVGD C0 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | ATP8B1 c.2083G>A p.(E695K) | Novel | AGVGD C55 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | PFIC type 1 | C; H | PEBD at 7 m C resolved |
| 29 | ATP8B1 c.2788C>T p.(R930*) | [54] (rs140407614) | Nonsense mutation predicted to result in nonsense-mediated decay | ATP8B1 c.2788C>T p.(R930*) | [54] (rs140407614) | Nonsense mutation predicted to result in nonsense-mediated decay | PFIC type 1 | C | LT Current symptoms unknown |
| 213 | ABCB11 c.731_732insA p.(I245Tfs*26) | Novel | Nonsense mutation predicted to result in nonsense-mediated decay | ABCB11 c.779G>A p.(G260D) | Novel | AGVGD C0 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | PFIC type 2 | C; H | PEBD at 14 m Pruritis resolved C persists |
| 130 | ABCB11 c.1081C>T p.(Q361*) | Novel | Nonsense mutation predicted to result in nonsense-mediated decay | ABCB11 c.1445A>G p.(D482G) | [23] (rs72549402) | AGVGD C65 SIFT Deleterious PP Probably damaging SSF No changes MES Cryptic donor NNS No changes GS No changes HSF Cryptic donor | PFIC type 2 | C; S | PEBD at 14 m C persists |
| 173 | ABCB11 c.1084-2A>G | Novel | SSF acceptor destroyed; cryptic acceptor MES acceptor destroyed; cryptic acceptor NNS acceptor destroyed; cryptic acceptor GS acceptor destroyed; cryptic acceptor HSF acceptor destroyed | ABCB11 c.1084-2A>G | Novel | SSF acceptor destroyed; cryptic acceptor MES acceptor destroyed; cryptic acceptor NNS acceptor destroyed; cryptic acceptor GS acceptor destroyed; cryptic acceptor HSF acceptor destroyed | PFIC type 2 | C; S; H; ALF | Not available |
| 93 | ABCB11 c.1409G>A p.(R470Q) | [55] | AGVGD C35 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | ABCB11 c.1409G>A p.(R470Q) | [55] | AGVGD C35 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | PFIC type 2 | C; H | LT Asymptomatic post-transplant |
| 180 | ABCB11 c.1409G>A p.(R470Q) | [55] | AGVGD C35 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | ABCB11 c.1409G>A p.(R470Q) | [55] | AGVGD C35 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | PFIC type 2 | C; S; H | Not available |
| 74 | ABCB11 c.1416T>A p.(Y472*) | [56] | Nonsense mutation predicted to result in nonsense-mediated decay | ABCB11 c.1416T>A p.(Y472*) | [56] | Nonsense mutation predicted to result in nonsense-mediated decay | PFIC type 2 | C; H; ALF | Not available |
| 97 | ABCB11 c.1445A>G p.(D482G) | [23] (rs72549402) | AGVGD C65 SIFT Deleterious PP Probably damaging SSF No changes MES Cryptic donor NNS No changes GS No changes HSF Cryptic donor | ABCB11 c.1445A>G p.(D482G) | [23] (rs72549402) | AGVGD C65 SIFT Deleterious PP Probably damaging SSF No changes MES Cryptic donor NNS No changes GS No changes HSF Cryptic donor | PFIC type 2 | C; S; H | PEBD at 9 m C resolved |
| 65 | ABCB11 c.1676T>C p.(M559T) | Novel | AGVGD C0 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS No changes HSF Cryptic donor | ABCB11 c.3933C>G p.(Y1311*) | Novel | Nonsense mutation predicted to result in nonsense-mediated decay | PFIC type 2 | C; S; H; ALF | LT Asymptomatic post-transplant |
| 209 | ABCB11 c.2708T>G p.(V903G) | Novel | AGVGD C35 SIFT Deleterious PP Benign SSF No changes MES No changes NNS No changes GS No changes HSF No changes | ABCB11 c.2708T>G p.(V903G) | Novel | AGVGD C35 SIFT Deleterious PP Benign SSF No changes MES No changes NNS No changes GS No changes HSF No changes | PFIC type 2 | C; S; H; ALF | Not available |
| 26 | ABCB11 c.3517A>G p.(N1173D) | [57] | AGVGD C0 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | ABCB11 c.3628A>C p.(T1210F) | [55,58,59] | AGVGD C0 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | PFIC type 2 | C | LT Asymptomatic post-transplant |
| 195 | ABCB11 c.3904G>T p.(E1302*) | [55] | Nonsense mutation predicted to result in nonsense-mediated decay | ABCB11 c.3904G>T p.(E1302*) | [55] | Nonsense mutation predicted to result in nonsense-mediated decay | PFIC type 2 | C; S; H | Not available |
| 168 | ABCB4 c.1230+1G>T | Novel | SSF donor destroyed MES donor destroyed NNS donor destroyed GS donor destroyed HSF donor destroyed | ABCB4 c.1230+1G>T | Novel | SSF donor destroyed MES donor destroyed NNS donor destroyed GS donor destroyed HSF donor destroyed | PFIC type 3 | C; S; H; ALF | Not available |
| 204 | ABCB4 c.1624G>C p.(A542P) | Novel | AGVGD C25 SIFT Deleterious PP Possibly damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | ABCB4 c.1624G>C p.(A542P) | Novel | AGVGD C25 SIFT Deleterious PP Possibly damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | PFIC type 3 | C; H | Not available |
| 36 | ABCB4 c.1652C>T p.(P551L) | Novel | AGVGD C65 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | ABCB4 c.1652C>T p.(P551L) | Novel | AGVGD C65 SIFT Deleterious PP Probably damaging SSF No changes MES No changes NNS No changes GS No changes HSF No changes | PFIC type 3 | C; H; ALF | LT Asymptomatic post-transplant |
| 162 | ABCB4 c.1858_1860delAAG p.(K620del) | Novel | SSF No changes MES No changes NNS No changes GS No changes HSF No changes | ABCB4 c.1858_1860delAAG p.(K620del) | Novel | SSF No changes MES No changes NNS No changes GS No changes HSF No changes | PFIC type 3 | H | Not available |
| Gene | Mutation | Protein Prediction Tools | Splicing Prediction Tools | Novel or Reference | Patient | Presenting Features | Final Diagnosis and Status at Follow Up |
|---|---|---|---|---|---|---|---|
| NPC1 | c.467T>C p.(M156T) | AGVGD C25 SIFT Deleterious PP benign | SSF No changes MES No changes NNS No changes GS No changes HSF No changes | (rs147615070) | 104 | C; H; S | Symptoms resolved No intervention Alternative diagnosis: multisystem juvenile xanthogranuloma |
| NPC1 | c.873G>T p.(W291C) | AGVGD C15 SIFT Deleterious PP Probably damaging | SSF No changes MES No changes NNS No changes GS No changes HSF No changes | (rs138151007) | 52 | C | Symptoms resolved No intervention |
| NPC1 | c.2010C>A p.(C670*) | Nonsense mutation predicted to result in nonsense-mediated decay | – | Novel | 4 | C; H | Progressive liver disease Alternative diagnosis: BA LT (22 m) Asymptomatic post-transplant |
| NPC1 | c.2010C>A p.(C670*) | Nonsense mutation predicted to result in nonsense-mediated decay | – | Novel | 10 | C; S | Symptoms resolved No intervention Alternative diagnosis: Hirschsprung’s disease necessitating parenteral nutrition. |
| NPC1 | c.3107C>T p.(T1036M) | AGVGD C15 SIFT Deleterious PP Probably damaging | SSF No changes MES No changes NNS No changes GS No changes HSF No changes | [60] (rs28942104) | 103 | C; H | Symptoms resolved No intervention |
| NPC1 | c.3614C>T p.(T1205I) | AGVGD C65 SIFT Deleterious PP Probably damaging | SSF Cryptic acceptor MES No changes NNS No changes GS No changes HSF No changes | Novel | 47 | C; H | Progressive liver disease with portal hypertension |
| NPC1 | c.3614C>T p.(T1205I) | AGVGD C65 SIFT Deleterious PP Probably damaging | SSF Cryptic acceptor MES No changes NNS No changes GS No changes HSF No changes | Novel | 71 | C; H; ALF | Progressive liver disease LT Asymptomatic post-transplant |
| ATP8B1 | c.287C>G p.(A96G) | AGVGD C0 SIFT Deleterious PP Possibly damaging | SSF No changes MES No changes NNS No changes GS No changes HSF No changes | Novel | 54 | C; H | Symptoms resolved. |
| ATP8B1 | c.2425A>C p.(I809L) | AGVGD C0 SIFT Deleterious PP Probably damaging | SSF No changes MES No changes NNS No changes GS No changes HSF No changes | Novel | 12 | C | Progressive liver disease Alternative diagnosis: BA LT (15 m) Asymptomatic post-transplant |
| ATP8B1 | c.3043T>C p.(F1015L) | AGVGD C15 SIFT Deleterious PP Probably damaging | SSF No changes MES No changes NNS No changes GS No changes HSF No changes | Novel | 32 | C; H; S; ALF | Progressive Liver disease Alternative diagnosis: BA LT (10 m) Asymptomatic post-transplant |
| ATP8B1 | c.3633C>A p.(F1211L) | AGVGD C15 SIFT Deleterious PP Probably damaging | SSF No changes MES No changes NNS No changes GS No changes HSF No changes | Novel | 5 | C | Symptoms resolved No intervention |
| ATP8B1 | c.3656A>G p.(D1219G) | AGVGD C65 SIFT Deleterious PP Probably damaging | SSF No changes MES No changes NNS No changes GS No changes HSF No changes | Novel | 15 | C | Progressive liver disease LT(30 m) Asymptomatic post-transplant |
| ABCB11 | c.1445A>G p.(D482G) | AGVGD C65 SIFT Deleterious PP Probably damaging | SSF No changes MES Cryptic donor NNS No changes GS No changes HSF Cryptic donor | [23] (rs72549402) | 206 | C; ALF | PEBD at 5 m Symptoms resolved |
| ABCB11 | c.1558A>T p.(R520*) | Nonsense mutation predicted to result in nonsense-mediated decay | – | 100 | C; H; S | Symptoms resolved No intervention | |
| ABCB4 | c.3317A>G p.(E1106G) | AGVGD C0 SIFT Deleterious PP Possibly damaging | SSF Cryptic acceptor MES Cryptic acceptor NNS No changes GS No changes HSF No changes | [23] (rs139042803) | |||
| ABCB11 | c.1621A>C p.(I541L) | AGVGD C0 SIFT Deleterious PP Probably damaging | SSF No changes MES No changes NNS No changes GS No changes HSF No changes | [58,61] | 208 | C | Symptoms resolved No intervention |
| ABCB11 | c.2678C>T p.(A893V) | AGVGD C0 SIFT Deleterious PP Probably damaging | SSF No changes MES No changes NNS No changes GS No changes HSF No changes | Novel | 190 | C; H; S | Not available |
| ABCB4 | c.524C>T p.(T175M) | AGVGD C65 SIFT Deleterious PP Probably damaging | SSF Cryptic acceptor MES No changes NNS No changes GS No changes HSF No changes | Novel | 181 | C; H | Not available |
| ABCB4 | c.1529A>G p.(N510S) | AGVGD C0 SIFT Deleterious PP Possibly damaging | SSF Cryptic donor MES No changes NNS No changes GS No changes HSF Cryptic acceptor | [62] (rs375315619) | 13 | C; S | Progressive liver disease Alternative diagnosis: BA LT (8 m) Asymptomatic post-transplant |
| ABCB4 | c.3403G>A p.(E1135K) | AGVGD C55 SIFT Deleterious PP Benign | SSF No changes MES No changes NNS No changes GS No changes HSF No changes | Novel | 53 | C; S; ALF | Progressive liver disease (multi-organ failure) Died age 15 m |
| SLC25A13 | c.1903G>T p.(D635Y) | AGVGD C15 SIFT Deleterious PP Possibly damaging | SSF No changes MES No changes NNS No changes GS No changes HSF No changes | Novel | 151 | C; H; S | Symptoms resolved No intervention |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeyaraj, R.; Bounford, K.M.; Ruth, N.; Lloyd, C.; MacDonald, F.; Hendriksz, C.J.; Baumann, U.; Gissen, P.; Kelly, D. The Genetics of Inherited Cholestatic Disorders in Neonates and Infants: Evolving Challenges. Genes 2021, 12, 1837. https://doi.org/10.3390/genes12111837
Jeyaraj R, Bounford KM, Ruth N, Lloyd C, MacDonald F, Hendriksz CJ, Baumann U, Gissen P, Kelly D. The Genetics of Inherited Cholestatic Disorders in Neonates and Infants: Evolving Challenges. Genes. 2021; 12(11):1837. https://doi.org/10.3390/genes12111837
Chicago/Turabian StyleJeyaraj, Rebecca, Kirsten McKay Bounford, Nicola Ruth, Carla Lloyd, Fiona MacDonald, Christian J. Hendriksz, Ulrich Baumann, Paul Gissen, and Deirdre Kelly. 2021. "The Genetics of Inherited Cholestatic Disorders in Neonates and Infants: Evolving Challenges" Genes 12, no. 11: 1837. https://doi.org/10.3390/genes12111837
APA StyleJeyaraj, R., Bounford, K. M., Ruth, N., Lloyd, C., MacDonald, F., Hendriksz, C. J., Baumann, U., Gissen, P., & Kelly, D. (2021). The Genetics of Inherited Cholestatic Disorders in Neonates and Infants: Evolving Challenges. Genes, 12(11), 1837. https://doi.org/10.3390/genes12111837