
The Mutational Landscape of PTK7 in Congenital Scoliosis and Adolescent Idiopathic Scoliosis

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Subjects
2.2. Bioinformatic Analysis and Variant Interpretation
- (1)
- Predicted to alter the protein sequence;
- (2)
- Either absent or with a low frequency (<0.001) from the public database mentioned above.
- (3)
- The missense variants have a CADD score ≥15.
2.3. Site-Directed Mutagenesis
2.4. Cell Culture and Transfection Assay
2.5. Western Blot Assay
2.6. Immuno-Fluorescence
2.7. Statistical Analysis
3. Results
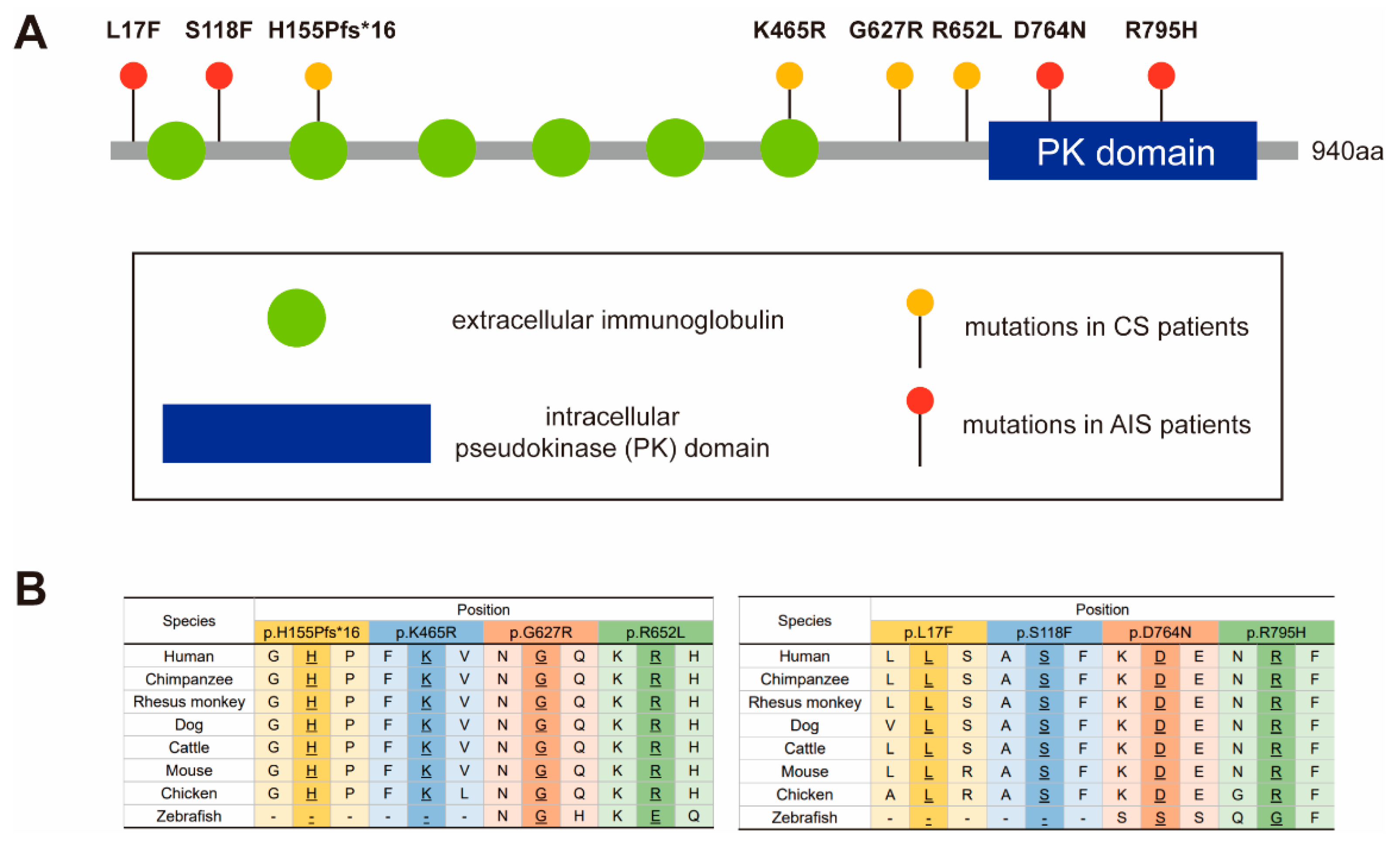
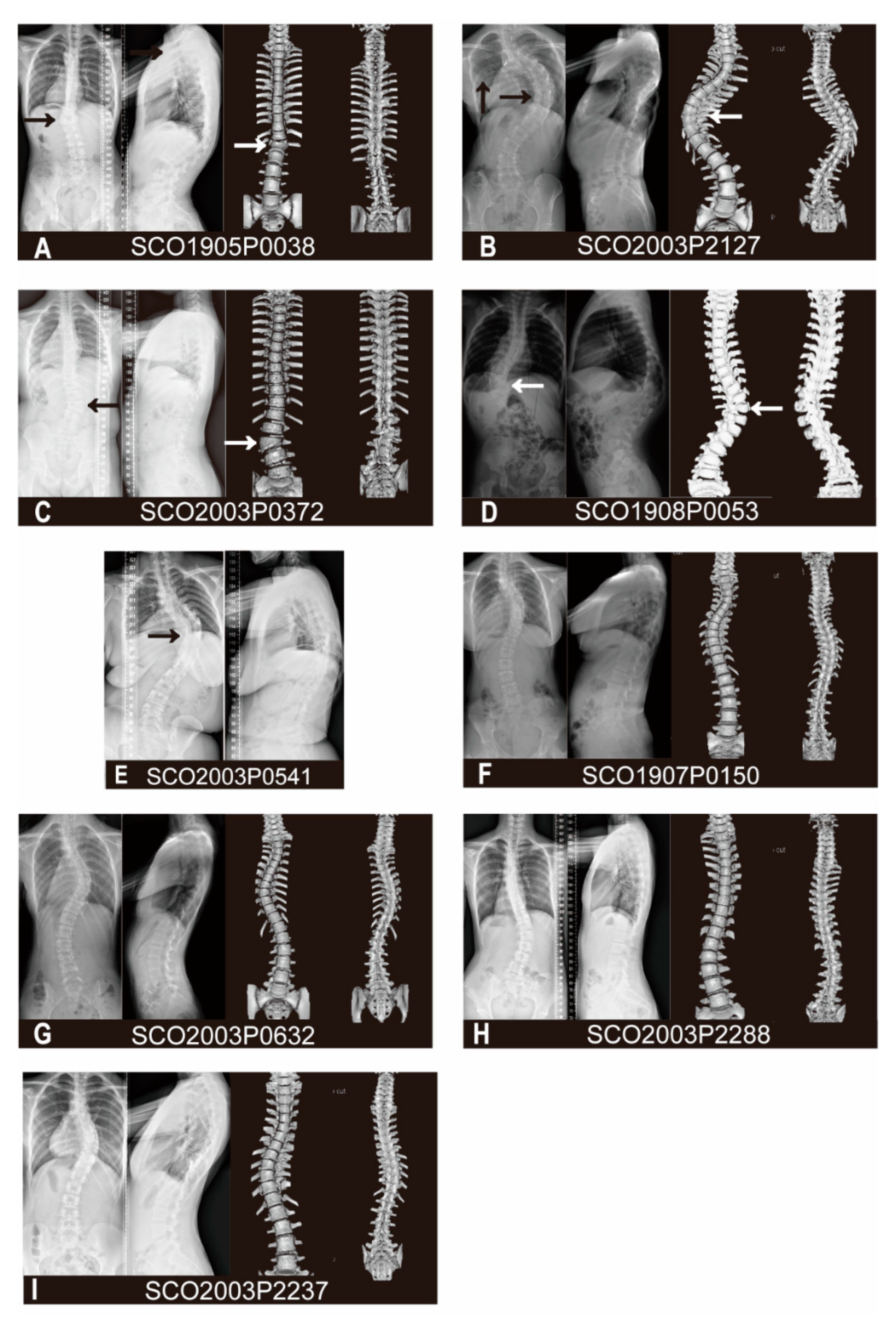
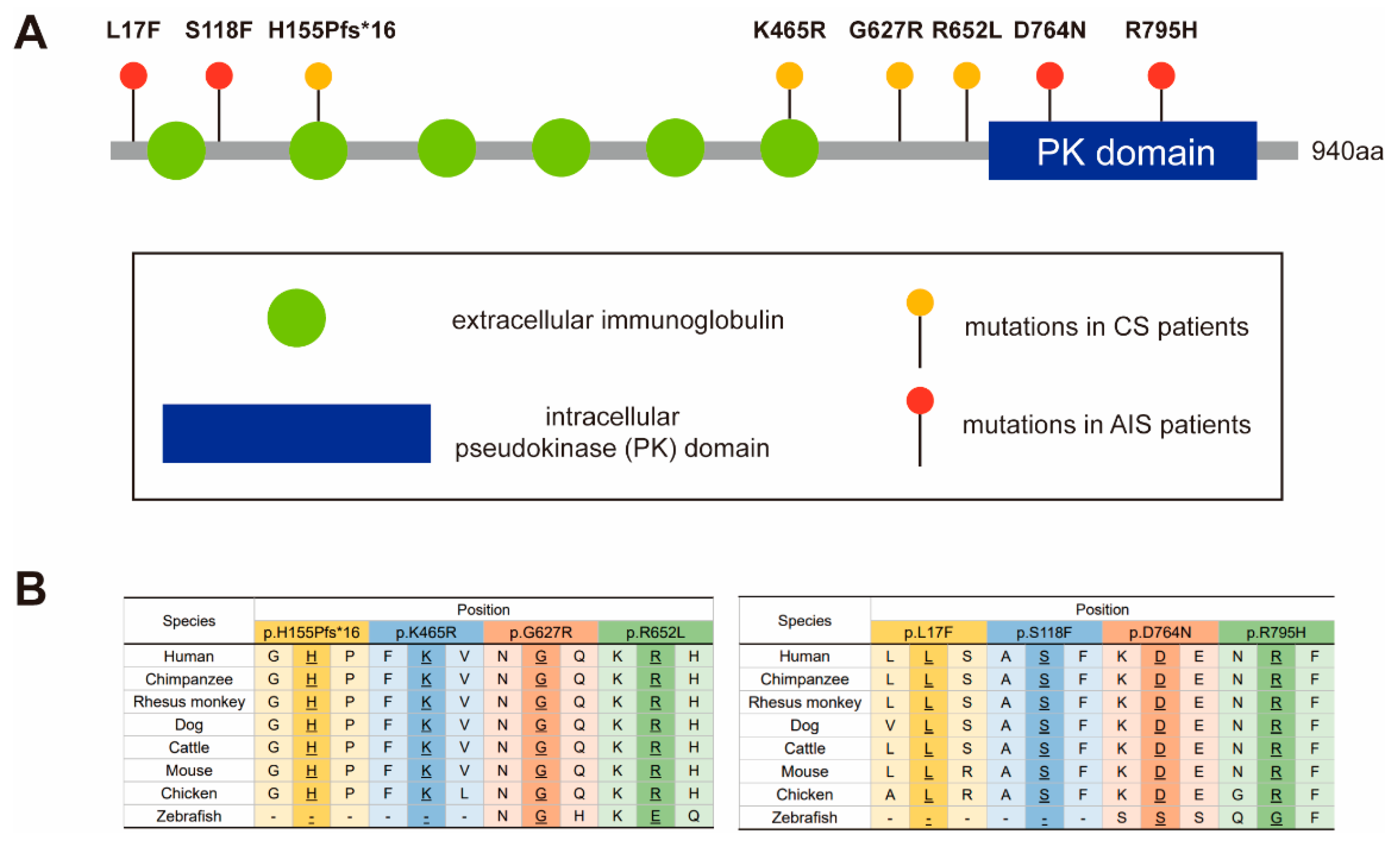
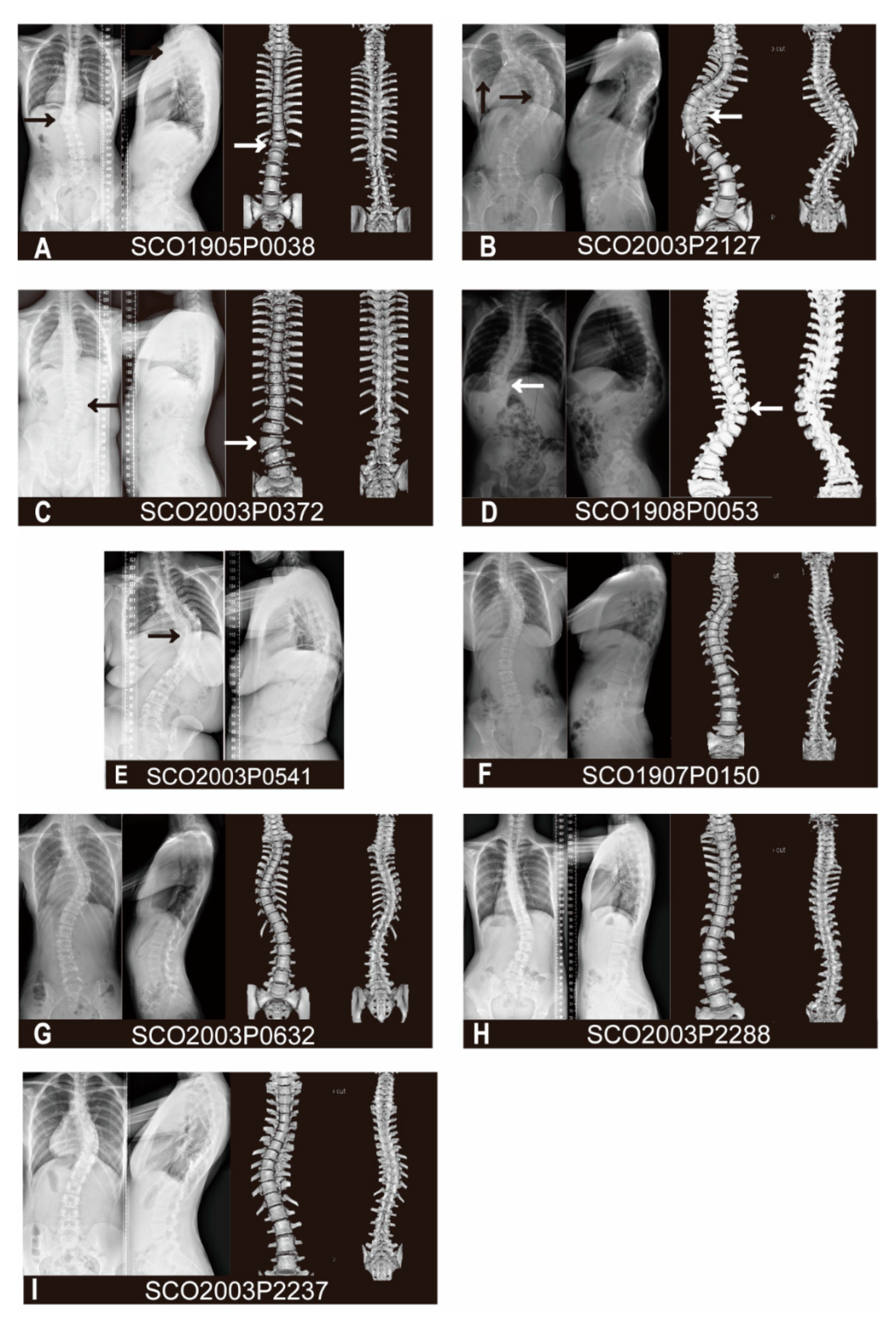
3.1. Variant and Phenotypic Characteristics
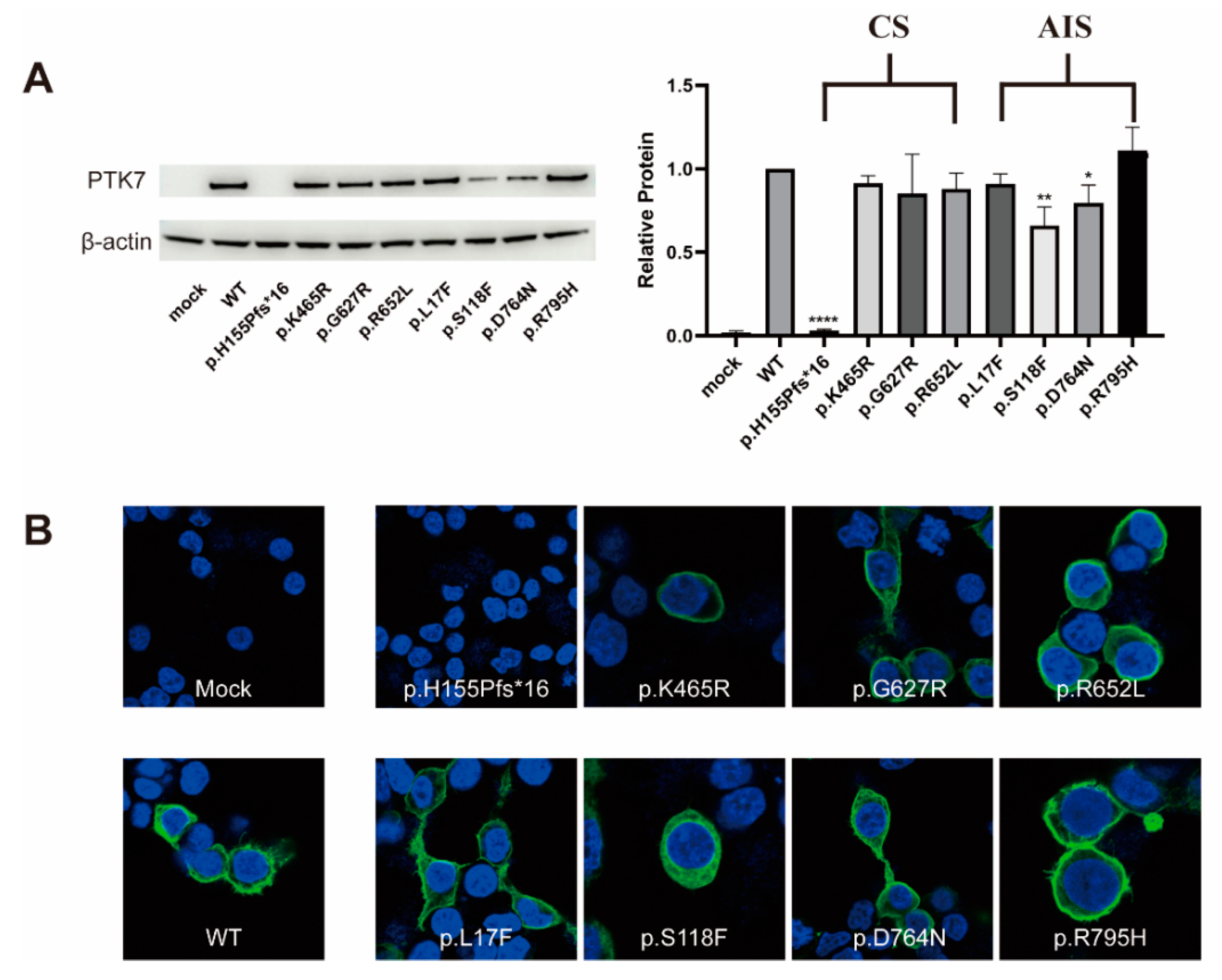
3.2. Western Blot and Immunocytochemistry Analyses
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mossie, K.; Jallal, B.; Alves, F.; Sures, I.; Plowman, G.D.; Ullrich, A. Colon carcinoma kinase-4 defines a new subclass of the receptor tyrosine kinase family. Oncogene 1995, 11, 2179–2184. [Google Scholar]
- Hayes, M.; Naito, M.; Daulat, A.; Angers, S.; Ciruna, B. Ptk7 promotes non-canonical Wnt/PCP-mediated morphogenesis and inhibits Wnt/beta-catenin-dependent cell fate decisions during vertebrate development. Development 2013, 140, 1807–1818. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.K.; Chauhan, S.K.; Kay, E.; Dana, R. Flt-1 regulates vascular endothelial cell migration via a protein tyrosine kinase-7-dependent pathway. Blood 2011, 117, 5762–5771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Borchers, A.G.; Jolicoeur, C.; Rayburn, H.; Baker, J.C.; Tessier-Lavigne, M. PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature 2004, 430, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.; Peradziryi, H.; Wehner, P.; Borchers, A. PlexinA1 interacts with PTK7 and is required for neural crest migration. Biochem. Biophys. Res. Commun. 2010, 402, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Capparuccia, L.; Tamagnone, L. Semaphorin signaling in cancer cells and in cells of the tumor microenvironment--two sides of a coin. J. Cell Sci. 2009, 122, 1723–1736. [Google Scholar] [CrossRef] [Green Version]
- Gay, C.M.; Zygmunt, T.; Torres-Vazquez, J. Diverse functions for the semaphorin receptor PlexinD1 in development and disease. Dev. Biol. 2011, 349, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Neufeld, G.; Kessler, O. The semaphorins: Versatile regulators of tumour progression and tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 632–645. [Google Scholar] [CrossRef]
- Moon, R.T.; Brown, J.D.; Torres, M. WNTs modulate cell fate and behavior during vertebrate development. Trends Genet. 1997, 13, 157–162. [Google Scholar] [CrossRef]
- Wodarz, A.; Nusse, R. Mechanisms of Wnt signaling in development. Annu. Rev. Cell Dev. Biol. 1998, 14, 59–88. [Google Scholar] [CrossRef] [Green Version]
- Hayes, M.; Gao, X.; Yu, L.X.; Paria, N.; Henkelman, R.M.; Wise, C.A.; Ciruna, B. ptk7 mutant zebrafish models of congenital and idiopathic scoliosis implicate dysregulated Wnt signalling in disease. Nat. Commun. 2014, 5, 4777. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Zhang, Y.; Chen, W.; Li, W.; Wang, S.; Wang, L.; Zhao, Y.; Lin, M.; Ye, Y.; Lin, J.; et al. Diagnostic yield and clinical impact of exome sequencing in early-onset scoliosis (EOS). J. Med. Genet. 2021, 58, 41–47. [Google Scholar] [CrossRef]
- Chen, N.; Zhao, S.; Jolly, A.; Wang, L.; Pan, H.; Yuan, J.; Chen, S.; Koch, A.; Ma, C.; Tian, W.; et al. Perturbations of genes essential for Mullerian duct and Wolffian duct development in Mayer-Rokitansky-Kuster-Hauser syndrome. Am. J. Hum. Genet. 2021, 108, 337–345. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, W.; O’Connor, T.D.; Jun, G.; Kang, H.M.; Abecasis, G.; Leal, S.M.; Gabriel, S.; Rieder, M.J.; Altshuler, D.; Shendure, J.; et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 2013, 493, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Kim, S.E.; Chen, Z.; Cao, X.; Zhu, H.; Yang, W.; Shaw, G.M.; Zheng, Y.; Zhang, T.; Wang, H.Y.; et al. Variants identified in PTK7 associated with neural tube defects. Mol. Genet. Genom. Med. 2019, 7, e00584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimes, D.T.; Boswell, C.W.; Morante, N.F.; Henkelman, R.M.; Burdine, R.D.; Ciruna, B. Zebrafish models of idiopathic scoliosis link cerebrospinal fluid flow defects to spine curvature. Science 2016, 352, 1341–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paudyal, A.; Damrau, C.; Patterson, V.L.; Ermakov, A.; Formstone, C.; Lalanne, Z.; Wells, S.; Lu, X.; Norris, D.P.; Dean, C.H.; et al. The novel mouse mutant, chuzhoi, has disruption of Ptk7 protein and exhibits defects in neural tube, heart and lung development and abnormal planar cell polarity in the ear. BMC Dev. Biol. 2010, 10, 87. [Google Scholar] [CrossRef] [Green Version]
- Berger, H.; Wodarz, A.; Borchers, A. PTK7 Faces the Wnt in Development and Disease. Front. Cell Dev. Biol. 2017, 5, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Mo, C.; Fraser, S.E. Planar cell polarity signalling controls cell division orientation during zebrafish gastrulation. Nature 2004, 430, 689–693. [Google Scholar] [CrossRef]
- Roszko, I.; Sawada, A.; Solnica-Krezel, L. Regulation of convergence and extension movements during vertebrate gastrulation by the Wnt/PCP pathway. Semin. Cell Dev. Biol. 2009, 20, 986–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Happe, H.; de Heer, E.; Peters, D.J. Polycystic kidney disease: The complexity of planar cell polarity and signaling during tissue regeneration and cyst formation. Biochim. Biophys. Acta 2011, 1812, 1249–1255. [Google Scholar] [CrossRef] [Green Version]
- Jessen, J.R.; Topczewski, J.; Bingham, S.; Sepich, D.S.; Marlow, F.; Chandrasekhar, A.; Solnica-Krezel, L. Zebrafish trilobite identifies new roles for Strabismus in gastrulation and neuronal movements. Nat. Cell Biol. 2002, 4, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Golubkov, V.S.; Strongin, A.Y. Downstream signaling and genome-wide regulatory effects of PTK7 pseudokinase and its proteolytic fragments in cancer cells. Cell Commun. Signal. 2014, 12, 15. [Google Scholar] [CrossRef] [Green Version]
- Koehler, A.; Schlupf, J.; Schneider, M.; Kraft, B.; Winter, C.; Kashef, J. Loss of Xenopus cadherin-11 leads to increased Wnt/beta-catenin signaling and up-regulation of target genes c-myc and cyclin D1 in neural crest. Dev. Biol. 2013, 383, 132–145. [Google Scholar] [CrossRef] [Green Version]
- Martinez, S.; Scerbo, P.; Giordano, M.; Daulat, A.M.; Lhoumeau, A.C.; Thome, V.; Kodjabachian, L.; Borg, J.P. The PTK7 and ROR2 Protein Receptors Interact in the Vertebrate WNT/Planar Cell Polarity (PCP) Pathway. J. Biol. Chem. 2015, 290, 30562–30572. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Patient Number | Variant | Gender | Age of Onset | Diagnosis | Vertebral Malformations | Other skeletal Malformations or Deformities | Cardiac Abnormalities | Spinal Canal Deformities | Visceral Abnormalities |
|---|---|---|---|---|---|---|---|---|---|
| SCO1905P0038 | c.464_465delAC | M | 13 | CS | T12 butterfly vertebra, T9-T10 failure of segmentation | None | None | None | None |
| SCO2003P2127 | c.1394A>G | F | 21 | CS | None | Fusion of 4th and 5th ribs, 12th ribs absent | Postoperative of patent ductus arteriosus | None | None |
| SCO2003P0372 | c.1879G>A | F | 8 | CS | L2 Hemivertebra, L2-L3 fusion | None | None | None | None |
| SCO1908P0053 | c.1955G>T | F | 1 | CS | T11, T12 segmented wedge vertebrae | None | None | None | None |
| SCO2003P0541 | c.1955G>T | F | 7 | CS | T7-T11 failure of segmentation | Chest deformity | None | Diastematomyelia | None |
| SCO1907P0150 | c.49C>T | F | 11 | AIS | None | None | Ventricular septal defect | None | None |
| SCO2003P0632 | c.353C>T | F | 12 | AIS | None | None | None | None | None |
| SCO2003P2288 | c.2290G>A | F | 14 | AIS | None | None | None | None | None |
| SCO2003P2237 | c.2384G>A | F | 13 | AIS | None | None | None | None | None |
| Patient Number | cDNA Change | Protein Change | Variant Type | Position | Gnomad_Exome_ALL | Gnomad_Exome_EAS | Gnomad_Genome_ALL | Gnomad_Genome_EAS | CADD | Ployphen-2 HDV Score | Evidence of Pathogenicity by ACMG | ACMG Classification |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SCO1905P0038 | c.464_465delAC | p.H155Pfs*16 | Frameshift | 43097560 | 0 | 0 | 0 | 0 | NA | NA | PVS1+PM2+PP3 | Pathogenic |
| SCO2003P2127 | c.1394A>G | p.K465R | Missense | 43109684 | 0 | 0 | 0.00003247 | 0.0006 | 26.2 | 0.997 | PP3 | Uncertain significance |
| SCO2003P0372 | c.1879G>A | p.G627R | Missense | 43112206 | 0.00004873 | 0.0006 | 0.00003232 | 0.0006 | 26.9 | 0.986 | PP3 | Uncertain significance |
| SCO1908P0053 SCO2003P0541 | c.1955G>T | p.R652L | Missense | 43112282 | 0.00007718 | 0.0005 | 0 | 0 | 23.6 | 0.947 | PP3 | Uncertain significance |
| SCO1907P0150 | c.49C>T | p.L17F | Missense | 43044275 | 0 | 0 | 0 | 0 | 19.95 | 0.997 | PM2 | Uncertain significance |
| SCO2003P0632 | c.353C>T | p.S118F | Missense | 43096988 | 0 | 0 | 0 | 0 | 25.6 | 0.982 | PM2 | Uncertain significance |
| SCO2003P2288 | c.2290G>A | p.D764N | Missense | 43114395 | 0.00000814 | 0.000058 | 0 | 0 | 23.8 | 0.114 | PM1+PP3 | Uncertain significance |
| SCO2003P2237 | c.2384G>A | p.R795H | Missense | 43126607 | 0.00000406 | 0 | 0.00003231 | 0 | 30 | 0.986 | PM1+ +PP3 | Uncertain significance |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Z.; Yang, Y.; Wang, S.; Zhao, S.; Zhao, H.; Li, X.; Niu, Y.; Deciphering Disorders Involving Scoliosis and COmorbidities (DISCO) Study Group; Qiu, G.; Wu, Z.; et al. The Mutational Landscape of PTK7 in Congenital Scoliosis and Adolescent Idiopathic Scoliosis. Genes 2021, 12, 1791. https://doi.org/10.3390/genes12111791
Su Z, Yang Y, Wang S, Zhao S, Zhao H, Li X, Niu Y, Deciphering Disorders Involving Scoliosis and COmorbidities (DISCO) Study Group, Qiu G, Wu Z, et al. The Mutational Landscape of PTK7 in Congenital Scoliosis and Adolescent Idiopathic Scoliosis. Genes. 2021; 12(11):1791. https://doi.org/10.3390/genes12111791
Chicago/Turabian StyleSu, Zhe, Yang Yang, Shengru Wang, Sen Zhao, Hengqiang Zhao, Xiaoxin Li, Yuchen Niu, Deciphering Disorders Involving Scoliosis and COmorbidities (DISCO) Study Group, Guixing Qiu, Zhihong Wu, and et al. 2021. "The Mutational Landscape of PTK7 in Congenital Scoliosis and Adolescent Idiopathic Scoliosis" Genes 12, no. 11: 1791. https://doi.org/10.3390/genes12111791
APA StyleSu, Z., Yang, Y., Wang, S., Zhao, S., Zhao, H., Li, X., Niu, Y., Deciphering Disorders Involving Scoliosis and COmorbidities (DISCO) Study Group, Qiu, G., Wu, Z., Wu, N., & Zhang, T. J. (2021). The Mutational Landscape of PTK7 in Congenital Scoliosis and Adolescent Idiopathic Scoliosis. Genes, 12(11), 1791. https://doi.org/10.3390/genes12111791