Abstract
Despite several decades of intensive research on spirurine nematodes, molecular data on some of the main lineages are still absent, which makes taxonomic classification insufficiently resolved. In the present study, we sequenced the first complete mitogenome for the family Quimperiidae, belonging to P. sinensis (Spirurina: Quimperiidae), a parasite living in the intestines of snakehead (Ophiocephalus argus). The circular mitogenome is 13,874 bp long, and it contains the standard nematode gene set: 22 transfer RNAs, 2 ribosomal RNAs and 12 protein-coding genes. There are also two long non-coding regions (NCR), in addition to only 8 other intergenic regions, ranging in size from 1 to 58 bp. To investigate its phylogenetic position and study the relationships among other available Spirurina, we performed the phylogenetic analysis using Bayesian inference and maximum likelihood approaches by concatenating the nucleotide sequences of all 36 genes on a dataset containing all available mitogenomes of the suborder Spirurina from NCBI and compared with gene order phylogenies using the MLGO program. Both supported the closer relationship of Ascaridoidea to Seuratoidea than to Spiruroidea. Pingus formed a sister-group with the Cucullanus genus. The results provide a new insights into the relationships within Spirurina.
1. Introduction
Nematoda, also known as roundworms, is one of the largest phyla in the animal kingdom. They are pseudocoelomic animals, mostly with small, cylindrical bodies. Nematodes are among the most common, abundant and ecologically diverse animal groups. Free-living species inhabit almost every environment, and they are extremely abundant in soils and aquatic sediments, both freshwater and marine [1,2]. The phylogeny of nematodes depends on a limited number of morphological and ecolog-ical characteristics, which can be determined before using molecular data. Due to the extreme speciosity of this phylum, the poor resolution conferred by morphologic characters resulted in incongruent classification systems [3]. Thus, the identification and phylogeny of nematodes are more suitable used with molecular data. However, since 18S rRNA genetic marker has insufficient resolution for such a large phylum, further studies must employ markers with much higher resolution power. [4]. Complete mitogenomes are much better suited for the taxonomic identification and phylogenetic studies of nematodes than morphology or traditionally used molecular markers because of much higher resolution power [5,6,7,8].
Mitochondrial gene order, i.e., the order in which genes are arranged on the mitochondrial genome, is sometimes used for phylogenetic reconstruction [9,10] and confirmation of findings based on mitochondrial DNA sequences [11]. As a result, mitogenomic data have often proven capable of clarifying phylogenetic relationships where other types of data offered a poor resolution, or where there was discordance between morphological features and genetic data, Furthermore, in many metazoans, the mitogenome accumulates mutations rate faster than the nuclear genome, which makes mitogenome a reliable source of phylogenetic data for distinguishing closely related taxa [12,13]. While mitogenomic data also have limitations for evolutionary studies [14,15], mitochondrial phylogenomics and comparative mitogenomics (e.g., gene order) have often been used as a useful tool for clarifying phylogenetic relationships in a large number of diverse animal groups with their ancient evolutionary origin [16,17,18,19]. Further comparative analyses of major nematode clades poorly represented in terms of mitogenomes are needed to fully explore the mitogenomic gene order evolution.
Spirurina, the representative suborder of gastrointestinal or tissues parasites of all clades of vertebrates, contains infraorders Ascaridomorpha, Gnathostomatomorpha, Oxyuridomorpha, Rhigonematomorpha, Spiruromorpha and Dracunculoidea as incertae sedi in the recent classification system based on molecular data [3]. Although the Spirurina clade is stably supported by the SSU rRNA data [20,21], the phylogenetic relationships for Spirurina based on mitogenomes remain inconclusive, largely due to the lack of sequences from several key lineages, which confuses the relationships within the clade and with other clades [22]. P. sinensis (Spirurina: Quimperiidae) is a roundworm parasitizing the intestine and caecum of fishes of the genus Channa. Although its morphological characteristics are unambiguous, molecular data is still absent, so its molecular phylogeny remains unstudied. As there are currently no complete mitogenomes available for the family Quimperiidae (Chromadorea: Spirurina: Seuratoidea), this study aims to sequence and characterize the complete mitogenome of the Quimperiidae species P. sinensis. As there are currently no molecular records at all for this species, these data shall also facilitate the identification of this species for future studies. Another objective was to conduct comparative analyses with all available Spirurina mitogenomes to study the relationships of the Spirurina. In addition, we also conducted comparative analyses of gene orders among the Spirurina, with the goal of inferring the evolutionary history of gene orders. The study first reveals phylogenetic position of family Quimperiidae and advances our understanding of the relationships within Spirurina.
2. Materials and Methods
2.1. Specimen Collection
Parasitic nematodes were acquired post mortem from the intestine of snakehead (Channa argus) samples caught by fishermen in the Lake Xiannv in Xinyu city, Jiangxi Province (24°29′14″–30°04′41″ N; 113°34′36″–118°28′58″ E), China, on 5 August 2020 and rinsed several times with physiological saline to wash out tissue particles of the host. The parasite was identified morphologically under a stereomicroscope and a light microscope according to the morphological features described by Moravec [23]. To further confirm the identity using molecular data, we relied on the 18S gene, which is recognized as a suitable molecular tool for the identification of nematodes [24]. The gene was amplified using self-designed primers. The sequence exhibited a 97.42% similarity with Paraseuratum sp. (Spirurina: Quimperiidae) (GenBank: KP275686). The samples were stored in 99% ethanol and 4 °C.
2.2. DNA Extraction and Amplification
The DNA extraction from a single specimen using a TIANamp Genomic DNA Kit (Tiangen Biotech, Beijing, China), following the manufacturer’s recommended protocol, and stored at −20 °C. Primers were designed to match generally conserved regions of target genes and used to amplify short fragments of cox1, rrnL, nad5, rrnS, nad1, cytb and nad4 genes. Specific primers were designed based on conserved regions and used to amplify the remaining mitogenome sequence in several PCR reactions (Table 1). The total volume for PCR was 20 μL: 1 μL rTaq polymerase (250U, Takara), 10 μL 2 × PCR buffer (Mg2+, dNTP plus, Takara, Dalian, China), 0.6 μL of each primer, 7.4 μL ddH2O and 1 μL of DNA template. The PCR amplification involved in an initial denaturation at 94 °C for 2 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 1 min/kb; a final extension at 72 °C for 10 min was then accomplished. The PCR products were sequenced bi-directionally at Sangon Company (Shanghai, China) using the primer-walking strategy.

Table 1.
Primers used to amplify and sequence the mitochondrial genome of P. sinensis.
2.3. Genome Assembly
After quality-proofing of the obtained fragments, the complete mitogenomic sequence of P. sinensis was assembled manually and aligned comparing with mitogenomes of related species by DNAstar v7.1 software [25] in accordance with the methodology outlined by Zou et al. [26] and Zhang et al. [27,28,29]. Briefly, raw mitogenomic sequences were imported into the MITOS web server to conclude the near boundaries of genes. The precise positions of protein-coding genes (PCGs) were inferred by searching for ORFs (employing the genetic code 5, invertebrate mitochondrial) and further corroborated via a comparison with homologs in Geneious [30]. All tRNAs were identified using ARWEN [31], DOGMA [32] and putative secondary structures were predicted using MITOS [33]. The genome annotation, reported in a Word (Microsoft Office) document, was input into PhyloSuite software [34] to parse and abstract the data, as well as generate GenBank submission files and organization tables for the mitogenome. Codon usage and relative synonymous codon usage (RSCU) for twelve protein-encoding genes (PCGs) of P. sinensis and Cucullanus robustus (Cucullanidae; the only complete mitogenome from the superfamily Seuratoidea currently available in the GenBank) were computed and the RSCU figure drawn using PhyloSuite. Tandem repeats in the non-coding region (NCRs) were identified by Tandem Repeats Finder [35], and the secondary structures of tRNAs were predicted by MITOS. PAL2NAL web tool [36] was used to calculate non-synonymous (dN)/synonymous (dS) mutation rates for twelve PCGs of P. sinensis and C. robustus. Rearrangement events and pairwise comparisons of gene orders of twelve spirurine nematodes were studied with CREx web tool [37] using the breakpoint comparison measurement. Bright colors represent similar genomes and dark colors represent dissimilar gene orders.
2.4. Phylogenetic and Gene Order Analyses
For the phylogeny, all available mitogenomes belonging to the suborder Spirurina were downloaded from the GenBank, and left only one mitogenome per species. Furthermore, two Dorylaimia (Nematoda: Enoplea) species, Romanomermis culicivorax and Trichuris muris, were used as outgroups. PhyloSuite was used to abstract the sequences of 36 genes (12 PCGs, 2 rRNAs and 22 tRNAs). All GenBank files were rearranged to start with the cox1 gene using the Reorder function in the same program, and the program was also used to generate the comparative mitogenomic architecture table for P. sinensis and C. robustus. All subsequent phylogenetic analysis steps were also conducted using PhyloSuite and its plug-in programs. Genes were aligned using MAFFT [38]: the normal alignment mode was appropriate for the rRNAs or tRNAs, and the codon alignment mode was appropriate for the 12 PCGs. Subsequently, a single alignment was generated by concatenating the manually optimized alignments using PhyloSuite. Based on the Akaike information criterion, GTR+G model was selected by PartitionFinder2 [39] as the best-suited evolutionary model. Two different algorithms: maximum likelihood (ML) and Bayesian inference (BI) were used to accomplish phylogenetic analyses. Bayesian inference was performed with default settings in MrBayes 3.2.6 [40] and accompanied with 1 × 106 metropolis-coupled MCMC generations. The best-fit partitioning strategy and models for partitions for IQ-TREE [41] were inferred using the inbuilt functions of IQ-TREE and followed by phylogenetic reconstruction with 50,000 ultrafast replicates [42]. Gene orders were visualized and annotated by iTOL [43] with the help of PhyloSuite. A phylogenetic tree was reconstructed using the MLGO program [44] on the basis of the gene order dataset, with 1000 bootstrap replicates [45].
3. Results
3.1. Genome Organization and Nucleotide Composition
The circular mitogenome of P. sinensis was 13,874 bp in length (Figure 1). The annotated mitogenome has been submitted to GenBank with accession number MW971502. The mitogenome encoded 36 genes: 12 PCGs (nad1-6, nad4L, cox1-3, atp6 and cytb), 22 tRNA genes (one tRNA for each amino acid, apart from leucine and serine, each of which was encoded by two tRNA genes) and 2 rRNA genes (s-rRNA and l-rRNA). All genes are transcribed from the same DNA strand. The absence of the atp8 gene in accordance with the general features of nematode mitogenomes with the exception of Trichinella spiralis, in which the atp8 gene was found [46]. In comparison to other nematodes [47,48], the base composition of the total mitogenome for P. sinensis was biased toward T and G (A + T content of 68.7%; 46.9% T; 22.4% G; 21.8% A; 8.9% C) rather than A and T, but the strand bias is consistent with other nematodes with a negative GC-skew and positive AT-skew. In conclusion, the nucleotides of metazoan mitogenomes are not randomly distributed, and this nucleotide bias is often linked with unequal usage of synonymous codons [49].
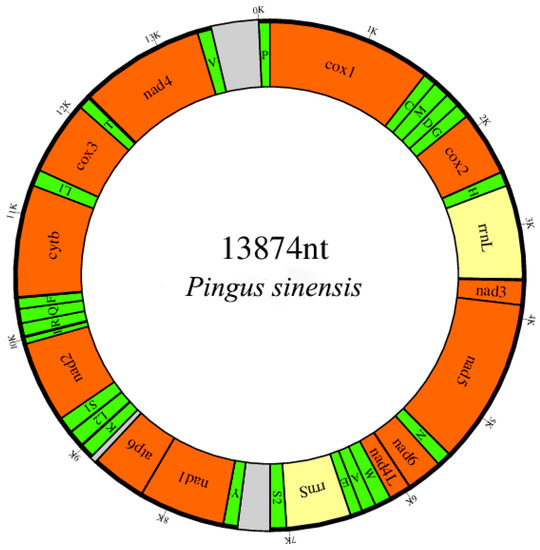
Figure 1.
Mitochondrial map of Pingus sinensis. The genes are denoted by the color blocks. PCGs are orange, rRNAs are yellow and tRNAs are green. The tRNAs are labelled according to the IUPACIUB single-letter amino acid code, with numerals distinguishing each of the two leucine- and serine-encoding tRNA genes (L1 and L2 for CUN and UUR, respectively; S1 and S2 for AGN and UCN, respectively), and intergenic regions are grey.
3.2. Protein-Coding Genes and Codon Usage
The twelve PCGs ranged in size from 234 bp (nad4L) to 1584 bp (nad5). Among the 12 PCGs, seven (nad1, nad2, nad3, nad6, cytb, cox1 and cox3) used TTG as the start codon, whereas four (nad4, nad4L, nad5, atp6) used ATT, and GTG was used only by the cox2 gene. TAA was the most commonly used termination codon (nad3, nad4, nad4L, nad5, nad6, cox3, atp6, cytb); three genes (cox1, cox2, nad1) used TAG, and the incomplete termination codon T-- was inferred only for the nad2 gene (Table 2). Same as C. robustus, the PCGs of the P. sinensis mitogenome are biased toward using amino acids encoded by T-, G- and A-rich codons. Codon usage, RSCU and codon family proportion (relevant to the amino acid usage) of P. sinensis and C. robustus are presented in Figure 2. Leucine (17.17%), phenylalanine (14.93%) and serine (10.22%) were the most frequent amino acids in the PCGs of P. sinensis, whereas arginine (0.97%), glutamine (1.33%) and histidine (1.5%) were relatively insufficient. The codons TTT (Phenylalanine, 14.3%) and TTG (Leucine, 8.6%) usage were most frequent, while the CGA codon for arginine was scarce (Additional file 1: Table S1). Therefore, codons ending in A or T were predominant, which consistent with the high A + T content of the third coding position of all PCGs in P. sinensis (Figure 2). The ratios of dN/dS for all twelve PCGs of P. sinensis against C. robustus ranged from 0.002 to 0.034 (Figure 3). All the PCGs were under negative (purifying) selection (ω < 1) pressure, indicating the existence of functional constraints affecting the evolution of these genes. Functional constraints on nad3, cytb, nad1 and nad6 genes were the most relaxed, whereas cox2 gene was evolving under the strongest purifying selection pressure (Figure 3).
Table 2.
The genome organization of P. sinensis.

Figure 2.
Relative Synonymous Codon Usage (RSCU) of P. sinensis and C. robustus. Values on the top of the bars refer to amino acid usage.
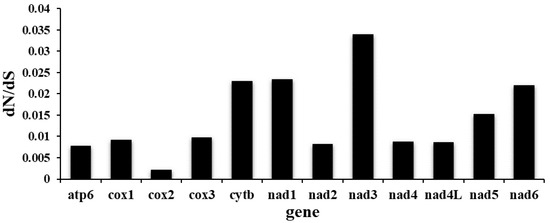
Figure 3.
Ratios of non-synonymous (dN)/synonymous (dS) nucleotide substitutions calculated from all 12 PCGs of P. sinensis and C. robustus mitogenomes.
3.3. Transfer RNA and Ribosomal RNA Genes
The small and large subunits of rrn (rrnS and rrnL) of P. sinensis were 682 bp and 945 bp in size, with 65.6% and 72.6% AT contents, respectively (Additional file 2: Table S2). All 22 commonly found tRNAs were identified in the mitogenome of P. sinensis, ranging from 52 bp (trnN (gtt)) to 62 bp (trnM (cat) and trnK (ttt)) in size, adding up to 1238 bp in length, with an average A + T content of 69.7% (Table 2). All 22 tRNA genes do not exhibit the standard cloverleaf secondary structure. The inferred structures of 20 of the twenty-two tRNA genes (except for two trnS genes) in P. sinensis consist of an amino-acyl stem of seven nucleotide pairs (ntp), a DHU-stem of 4 ntp with a 3–7 nt loop, an anticodon stem of 5 ntp with a loop, and a unique characteristic in that the TΨC arm and variable loop are replaced by a ‘TV replacement loop’ of 6–14 bases. The trnS possesses a secondary structure including a DHU replacement loop of 4 bases, a 5 bp TΨC arm, a TΨC loop of 4 bases and a variable loop of 3–4 bases. Noncanonical tRNA structure are common in nematodes, and secondary structures predicted for the transcripts of these genes are similar to those reported from a variety of other nematode mitogenomes [47,48,50,51,52,53,54], excluding T. spiralis in which some tRNAs have the canonical cloverleaf structures [40].
3.4. Non-Coding Regions
There were 8 short intergenic regions ranging from 1 to 58 bp in size, adding up to a total of 393 bp. In addition, there were also two long non-coding regions (>100 bp): NCR1 (between trnS2 and trnY) and NCR2 (between trnV and trnP). They were 284 bp and 417 bp long, with 61.3% and 60.4% AT content, respectively (Additional file 2: Table S2). Tandem repeats are inferred to result from strand slippage during replication [55], and they have been identified in large NCRs of nematodes, including CR1-CR6 in Caenorhabditis elegans and the dinucleotide repeat region in Ascaris suum [52]. However, the two NCRs of P. sinensis were absence of consecutive sequences of A and T, and we did not identify any tandem repeats.
3.5. Phylogeny
Phylogenetic relationships of P. sinensis with related nematodes of the Spirurina were presumed using ML and BI methods. The dataset comprised 36 concatenated genes (12 PCGs, 2 rRNAs and 22 tRNAs). Both methods, BI and ML, produced an identical topology, and most of the nodes exhibited high bootstrap resampling (ML) and posterior probability (BI) values. Additional file 3: Figure S1 shows the combined strict accordance phylogenetic tree produced from the two phylogenetic methods. As some superfamilies were represented by a single species in our study, this topology should be comprehended with some caution. Tree topology suggests the existence of two major clades: one clade includes the superfamilies Physalopteroidea, Thelazioidea, Spiruroidea and Filarioidea; another clade consists of the superfamilies Oxyuroidea, Camallanoidea, Dracunculoidea, Rhigonematoidea, Heterakoidea, Seuratoidea, Gnathostomatoidea and Ascaridoidea. The second clade was further sub-divided into two clades, Oxyuroidea and (Camallanoidea + (Dracunculoidea + (Rhigonematoidea + (Heterakoidea + (Seuratoidea + (Gnathostomatoidea + (Ascaridoidea))))))), and a monophyletic clade comprising (Physalopteridae + (Thelazioidea + (Gongylonematidae + (Setariidae + (Onchocercidae))))). Most clades were robustly supported. This relationship is in agreement with phylogenies inferred by Černotíková et al. [56] and Zou et al. [5]. However, our results appear to reject the hypothesis that Oxyuroidea represented the earliest branch of this clade [3,20], our results in agreement with a previous study [57].
The topology obtained in our phylogenetic analysis supports a sister relationship between Seuratoidea and Ascaridoidea, while strongly rejecting the Ascaridoidea-Spiruroidea affinity [47,50]. Meanwhile, the sister relationship also is supported between the Camallanoidea and Dracunculoidea. Ascarididae and Anisakidae were resolved as paraphyletic, along with the particularly problematic phylogeny of the Ortleppascaris genus, which appears to exhibit a closer relationship with the family Anisakidae than Ascarididae. Intriguingly, the Ophidascaris genus is closer to the family Ascarididae than Anisakidae. In our study, in addition, Gnathostomatoidea formed a sister clade with (Seuratoidea + Ascaridoidea), which is also incongruent with Zou et al. [5]. And the sister group relationship between the Quimperiidae family and Cucullanidae family was revealed, mitochondrial gene order and transformational pathway analysis provided additional supporting evidence for the close relationship between these two families in this study (Additional file 4: Figure S2).
3.6. GO Analysis
For a better comparison of gene orders among the Spirurina, Phylosuite was used to extract and visualize all available sequences from the NCBI. The gene order of P. sinensis is very similar to that of C. robustus, with only one transposition event: a position change of trnN; three TDRL operations are required to Gnathostoma doloresi; a single transposition event and two TDRL operations are required to Ophidascaris baylisi (Additional file 5: Figure S3). Such gene arrangements are interpreted to be important for molecular systematics and phylogenetic reconstruction because of the rarity of mitochondrial gene arrangement events [58]. Meanwhile, the mitochondrial gene orders of P. sinensis and C. robustus show that Seuratoidea species do not shows a synapomorphic gene order, and indicate that gene order within this group may be evolving at uneven rates.
In order to evaluate the phylogenetic power of gene order, we reconstructed a phylogeny using gene orders (MLGO algorithm), and compared it with the phylogeny inferred using mitogenomic sequences. The monophyly of (Physalopteroidea + (Spiruroidea + (Thelazioidea + (Filarioidea)))) was supported both by the gene order and mitogenomic sequences. Paraphyly of Camallanoidea and Oxyuroidea was also supported by both topologies, as was the sister-group relationship of Pingus and Cucullanus genera. There were also some differences: Onchocerca flexuosa, Chandlerella quiscali, Litomosoides sigmodontis and Ruizia karukerae, which have gene arrangements that do not conform to the gene orders of their closest relatives and topologies. Disregarding the tRNA genes, there were only 9 types of gene orders; this has been believed as additional evidence of their close relationship [6] (Additional file 6: Figure S4), tRNA genes possess small size which probably result in translocations accumulate more quickly. [59]. Hence, designating tRNA genes as hypermobile and excluding them from gene order phylogenies is a feasibile option when dealing with different rearrangement rates [60].
4. Discussion
In conclusion, our analysis reveals the phylogenetic position of family Quimperiidae and provides an insight into the phylogenetic relationships of Spirurina, although the availability of molecular data remains limited. We have sequenced and characterized the complete mitogenome of the fish-parasitizing spirurine nematode P. sinensis, which includes the standard 22 rRNA genes, 2 rRNA genes and 12 PCGs. There are no highly repetitive regions in the NCR. Using concatenated nucleotide sequences of all 36 genes, the phylogenetic analyses were conducted using Bayesian inference and maximum likelihood methods, providing insight into the relationships within the Spirurina. Meanwhile, gene order can be used with discretion as a source of phylogenetic data in nematodes. The monophyly of (Physalopteroidea + (Spiruroidea + (Thelazioidea + (Filarioidea)))) and paraphyly of Camallanoidea and Oxyuroidea are supported both by the gene order and mitogenomic sequences, as was the sister-group relationship of Pingus and Cucullanus genera. However, gene orders are inclined to produce artefactual relationships and should be accompanied with utmost caution for phylogenetic reconstruction in Nematoda. Further studies should centre around adding mitogenomes of other inadequately represented taxa for purpose of advancing understanding of the evolution and variation of nematode trophic ecologies and infer their evolutionary relationships with higher resolution.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/genes12111772/s1, Additional file 1: Table S1: Nucleotide codon usage for 12 protein-coding genes of the mitochondrial genome of P. sinensis. Additional file 2: Table S2: Nucleotide skewness and composition of the protein-coding genes, tRNAs and rRNAs of mitochondrial genomes of P. sinensis and C. robustus. Additional file 3: Figure S1: Phylogeny of 20 families deduced from 36 genes. Scale-bar indicates the estimated number of substitutions per site. The red star symbol represents that both methods generated a maximum statistical support value (BP = 100, BPP = 1.0); elsewhere ML/BI values are shown above the node. Taxonomic families and genes are shown in different colors. A dash (-) before the gene name represents that the gene is encoded on the minus strand. Additional file 4: Figure S2: Pairwise breakpoint comparison of gene orders among fourteen Spirurina species on the basis of the order of 36 genes. Scores represent the dissimilarity between gene orders, whereas ‘0’ indicates an identical gene order. Additional file 5: Figure S3: Transformational pathways from the gene order of P. sinensis to the most similar nematode gene arrangements. Additional file 6: Figure S4: Phylogenetic tree on the basis of the gene order dataset. Taxonomic identity (superfamilies or families) is shown on the left. A red dot on the clade nod represents the clades that have similar topologies with Figure 3.
Author Contributions
F.C. participated in the study conception and design, sample collection, data analysis, data visualization and manuscript preparation. H.Z. participated in the same sample collection, molecular lab work and data analysis. X.J. participated in the same molecular lab work. D.Z. participated in the same data analysis. W.L., M.L. and S.W. participated in the study conception and design. G.W. participated in the study conception and design and supervised the study. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Second Tibetan Plateau Scientific Expedition and Re-search Program (STEP) (No. 2019QZKK0304). The funders had no role in the design of the study, in the collection, analysis and interpretation of the data or in the writing the manuscript.
Institutional Review Board Statement
All experimental procedures involving animals were reviewed, approved and supervised by the Animal Care Committee of the Institute of Hydrobiology, Chinese Academy of Sciences. There are no special permits were required to retrieve and process the samples because of the study did not involve any live vertebrates, nor regulated invertebrates.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to thank Chen-Rong (Bio-Transduction Lab) for helping us to conduct some of the wet lab experiments and Ivan Jakovlić for manuscript polishing. We would also like to thank the editor and the two anonymous reviewers for the time they have invested in reviewing our manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Golden, A.M. Classification of the genera and higher categories of the order Tylenchida (Nematoda). J. Parasitol. 1970, 56, 191–232. [Google Scholar]
- Poinar, G.O. Nematoda and Nematomorpha. In Ecology and Classification of North American Freshwater Invertebrates; Thorp, J.H., Covich, A.P., Eds.; Academic Press: Cambridge, MA, USA, 2010; Chapter 9; pp. 237–276. [Google Scholar]
- De Ley, P.; Blaxter, M.L. Systematic Position and Phylogeny. In The Biology of Nematodes; Taylor & Francis: London, UK, 2002; pp. 1–30. [Google Scholar]
- Koutsovoulos, G.D. Reconstructing the Phylogenetic Relationships of Nematodes Using Draft Genomes and Transcriptomes: University of Edinburgh. Ph.D. Thesis, University of Edinburgh, Edinburgh, UK, 2015. [Google Scholar]
- Zou, H.; Jakovlić, I.; Chen, R.; Zhang, D.; Zhang, J.; Li, W.-X.; Wang, G.-T. The complete mitochondrial genome of parasitic nematode Camallanus cotti: Extreme discontinuity in the rate of mitogenomic architecture evolution within the Chromadorea class. BMC Genom. 2017, 18, 840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-K.; Sultana, T.; Lee, S.-H.; Kang, S.; Kim, H.K.; Min, G.-S.; Eom, K.; Nadler, S.A. Monophyly of clade III nematodes is not supported by phylogenetic analysis of complete mitochondrial genome sequences. BMC Genom. 2011, 12, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchêne, S.; Archer, F.I.; Vilstrup, J.; Caballero, S.; Morin, P.A. Mitogenome phylogenetics: The impact of using single regions and partitioning schemes on topology, substitution rate and divergence time estimation. PLoS ONE 2011, 6, e27138. [Google Scholar] [CrossRef] [Green Version]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boore, J.L. Big trees from little genomes: Mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef]
- Fritzsch, G.; Schlegel, M.; Stadler, P.F. Alignments of mitochondrial genome arrangements: Applications to metazoan phylogeny. J. Theor. Biol. 2006, 240, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb, C.A.; Saunders, N.C. Intraspecific Phylogeography: The Mitochondrial DNA Bridge Between Population Genetics and Systematics. Annu. Rev. Ecol. Syst. 1987, 18, 489–522. [Google Scholar] [CrossRef]
- Brown, W.M.; George, M.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef] [Green Version]
- Vawter, L.; Brown, W. Nuclear and mitochondrial DNA comparisons reveal extreme rate variation in the molecular clock. Science 1986, 234, 194–196. [Google Scholar] [CrossRef]
- Rubinoff, D.; Holland, B.S.; Savolainen, V. Between Two Extremes: Mitochondrial DNA is neither the Panacea nor the Nemesis of Phylogenetic and Taxonomic Inference. Syst. Biol. 2005, 54, 952–961. [Google Scholar] [CrossRef]
- Zhang, D.; Zou, H.; Hua, C.J.; Li, W.-X.; Mahboob, S.; Al-Ghanim, K.A.; Al-Misned, F.; Jakovlić, I.; Wang, G.-T. Mitochondrial Architecture Rearrangements Produce Asymmetrical Nonadaptive Mutational Pressures That Subvert the Phylogenetic Reconstruction in Isopoda. Genome Biol. Evol. 2019, 11, 1797–1812. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.J.; Arndt, A.; Gorski, S.; Fajber, E. The phylogeny of echinoderm classes based on mitochondrial gene arrangements. J. Mol. Evol. 1993, 36, 545–554. [Google Scholar] [CrossRef]
- Boore, J.L.; Brown, W.M. Mitochondrial genomes of Galathealinum, Helobdella, and Platynereis: Sequence and gene arrangement comparisons indicate that Pogonophora is not a phylum and Annelida and Arthropoda are not sister taxa. Mol. Biol. Evol. 2000, 17, 87–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, U.W.; Friedrich, M.; Tautz, D.; Park, C.J.; Kim, W. Mitochondrial protein phylogeny joins myriapods with chelicerates. Nature 2001, 413, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Bret, L.; Simon, D.L.; Kadane, J.B.; Deborah, S. A bayesian analysis of metazoan mitochondrial genome arrangements. Mol. Biol. Evol. 2005, 22, 486–495. [Google Scholar]
- Blaxter, M.L.; De Ley, P.; Garey, J.R.; Liu, L.X.; Scheldeman, P.; Vierstraete, A.; Vanfleteren, J.R.; Mackey, L.Y.; Dorris, M.; Frisse, L.M.; et al. A molecular evolutionary framework for the phylum Nematoda. Nature 1998, 392, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Blaxter, M.L. Nematoda: Genes, genomes and the evolution of parasitism. Adv. Parasitol. 2003, 54, 101–195. [Google Scholar]
- Wijová, M.; Moravec, F.; Horák, A.; Lukes, J. Evolutionary relationships of Spirurina (Nematoda: Chromadorea: Rhabditida) with special emphasis on dracunculoid nematodes inferred from SSU rRNA gene sequences. Int. J. Parasitol. 2006, 36, 1067–1075. [Google Scholar] [CrossRef]
- Moravec, F.; Nie, P.; Wang, G.; Moravec, F.; Nie, P.; Wang, G. Some nematodes of fishes from central China, with the redescription of Procamallanus (Spirocamallanus) fulvidraconis (Camallanidae). Folia Parasitol. 2003, 50, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floyd, R.M.; Rogers, A.D.; Lambshead, P.J.D.; Smith, C.R. Nematode-specific PCR primers for the 18S small subunit rRNA gene. Mol. Ecol. Notes 2005, 5, 611–612. [Google Scholar] [CrossRef]
- Burland, T.G. DNASTAR’s Lasergene sequence analysis software. In Bioinformatics Methods and Protocols; Humana Press: Totowa, NJ, USA, 2000; pp. 71–91. [Google Scholar]
- Zou, H.; Jakovli, I.; Zhang, D.; Hua, C.J.; Wang, G.T. Architectural instability, inverted skews and mitochondrial phylogenomics of Isopoda: Outgroup choice affects the long-branch attraction artefacts. R. Soc. Open Sci. 2020, 7, 191887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Li, W.X.; Zou, H.; Wu, S.G.; Li, M.; Jakovlić, I.; Zhang, J.; Chen, R.; Wang, G.T. Mitochondrial genomes of two diplectanids (Platyhelminthes: Monogenea) expose paraphyly of the order Dactylogyridea and extensive tRNA gene rearrangements. Parasites Vectors 2018, 11, 601. [Google Scholar] [CrossRef]
- Zhang, D.; Zou, H.; Jakovli, I.; Wu, S.G.; Li, M.; Zhang, J.; Chen, R.; Li, W.X.; Wang, G.T. Mitochondrial Genomes of Two Thaparocleidus Species (Platyhelminthes: Monogenea) Reveal the First rRNA Gene Rearrangement among the Neodermata. Int. J. Mol. Sci. 2019, 20, 4214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Zou, H.; Wu, S.G.; Li, M.; Li, W.X. Sequencing, characterization and phylogenomics of the complete mitochondrial genome of Dactylogyrus lamellatus (Monogenea: Dactylogyridae). J. Helminthol. 2017, 92, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2007, 24, 172–175. [Google Scholar] [CrossRef] [Green Version]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [Green Version]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2019, 20, 348–355. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernt, M.; Merkle, D.; Ramsch, K.; Fritzsch, G.; Perseke, M.; Bernhard, D.; Schlegel, M.; Stadler, P.F.; Middendorf, M. CREx: Inferring genomic rearrangements based on common intervals. Bioinformatics 2007, 23, 2957–2958. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.; Huelsenbeck, J. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Hu, F.; Lin, Y.; Tang, J. MLGO: Phylogeny reconstruction and ancestral inference from gene-order data. BMC Bioinform. 2014, 15, 354. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Hu, F.; Tang, J.; Moret, B.M.E. Maximum likelihood phylogenetic reconstruction from high-resolution whole-genome data and a tree of 68 eukaryotes. Biocomputing 2013, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Lavrov, D.V.; Brown, W.M. Trichinella spiralis mtDNA: A nematode mitochondrial genome that encodes a putative ATP8 and normally structured tRNAS and has a gene arrangement relatable to those of coelomate metazoans. Genetics 2001, 157, 621–637. [Google Scholar] [CrossRef]
- Kim, K.H.; Eom, K.S.; Park, J.K. The complete mitochondrial genome of Anisakis simplex (Ascaridida: Nematoda) and phylogenetic implications. Int. J. Parasitol. 2006, 36, 319–328. [Google Scholar] [CrossRef]
- Hu, M.; Chilton, N.B.; Gasser, R.B. The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea). Int. J. Parasitol. 2002, 32, 145–158. [Google Scholar] [CrossRef]
- Herbeck, J.; Novembre, J. Codon Usage Patterns in Cytochrome Oxidase I Across Multiple Insect Orders. J. Mol. Evol. 2003, 56, 691–701. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Chilton, N.B.; Gasser, R.B. The mitochondrial genome of Strongyloides stercoralis (Nematoda)—Idiosyncratic gene order and evolutionary implications. Int. J. Parasitol. 2003, 33, 1393–1408. [Google Scholar] [CrossRef]
- Unnasch, H.T.R. The mitochondrial genome of Onchocerca volvulus: Sequence, structure and phylogenetic analysis. Mol. Biochem. Parasitol. 1998, 95, 111–127. [Google Scholar]
- Okimoto, R.; Macfarlane, J.L.; Clary, D.O.; Wolstenholme, D.R. The Mitochondrial Genomes of Two Nematodes, Caenorhabditis elegans and Ascaris suum. Genetics 1992, 130, 471–498. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, A.; Small, S.T.; Kloos, Z.A.; Kazura, J.W.; Nutman, T.B.; Serre, D.; Zimmerman, P.A. The complete mitochondrial genome sequence of the filarial nematode Wuchereria bancrofti from three geographic isolates provides evidence of complex demographic history. Mol. Biochem. Parasitol. 2012, 183, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultana, T.; Kim, J.; Lee, S.H.; Han, H.; Park, J.K. Comparative analysis of complete mitochondrial genome sequences confirms independent origins of plant-parasitic nematodes. BMC Evol. Biol. 2013, 13, 12. [Google Scholar] [CrossRef] [Green Version]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar]
- Černotíková, E.; Horák, A.; Moravec, F. Phylogenetic relationships of some spirurine nematodes (Nematoda: Chromadorea: Rhabditida: Spirurina) parasitic in fishes inferred from SSU rRNA gene sequences. Folia Parasitol. 2011, 58, 135–148. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.H.; Jia, Y.Q.; Wang, Y.N.; Zhao, G.H.; Zhu, X.-Q. The complete mitochondrial genome of the gullet worm Gongylonema pulchrum: Gene content, arrangement, composition and phylogenetic implications. Parasites Vectors 2015, 8, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boore, J.L.; Fuerstenberg, S.I. Beyond linear sequence comparisons: The use of genome-level characters for phylogenetic reconstruction. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Kern, E.; Kim, T.; Park, J.K. The mitochondrial genome in nematode phylogenetics. Front. Ecol. Evol. 2020, 8, 250. [Google Scholar] [CrossRef]
- Kim, J.; Kern, E.; Kim, T.; Sim, M.; Kim, J.; Kim, Y.; Park, C.; Nadler, S.A.; Park, J.-K. Phylogenetic analysis of two Plectus mitochondrial genomes (Nematoda: Plectida) supports a sister group relationship between Plectida and Rhabditida within Chromadorea. Mol. Phylogenetics Evol. 2017, 107, 90–102. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).