1. Introduction
Next-generation sequencing (NGS) is now regularly used to identify viral sequences in the biological sample of an infected host in order to relate the presence of a virus with disease symptoms of the host [
1,
2,
3]. Besides detection of individual viruses, NGS data is regularly used to determine the overall taxonomic composition of pathogens for the purpose of comparative metagenomics [
4] or to reconstruct networks between microbial taxa [
5]. Most computational virus detection pipelines or pipelines for determining the taxonomic composition map sequencing reads or assembled contigs against viral reference sequences available in public or own curated databases [
6,
7,
8,
9,
10,
11,
12,
13]. These mapping approaches have been proven to be successful in a large number of examples, however, they mostly fail to classify reads from new emerging viruses whose sequences are not yet deposited in a database. Ren et al. [
13], for example, mention that novel viruses ‘for which their hallmark genes have not been characterized or are poorly represented in reference databases’ can be missed. The difficulty of identifying novel viruses when also no homologues are detectable in sequence databases was pointed out by Maclot et al. [
14]. Parks et al. [
15] argue that an effective taxonomic assignment algorithm ‘must deal explicitly with unrepresented lineages, either by mapping such novelty to an appropriate taxonomic level [...] or by using a criterion that allows some reads or assemblies to remain unassigned’. While there are diverse tools available for taxonomic classification of NGS reads from novel species, these share several limitations. For instance, the Kraken-1 tool [
16] matches
k-mers (with
k up to 31) with a reference of genome sequences; however, the sensitivity drops to
in the best case if the species is yet unknown. As the authors state, this is due to ‘Kraken’s reliance on exact matches of relatively long
k-mers: sequences deriving from different genera rarely share long exact matches’. Despite improvements of the metagenomic data analysis algorithm, Kraken-2 [
17] still shows low sensitivity on novel viruses.
Besides classification approaches by mapping, machine learning approaches have been used to classify reads to taxonomic ranks [
18] such as genus, family, order, class and phylum. Among the machine learning approaches listed by Peabody et al. [
18], only one approach studies the performance of deep learning models for taxonomic classification (Rasheed et al. [
19]). Rasheed et al. use artificial neural networks (ANN) to classify reads to different taxonomic ranks and evaluate their models on datasets representing about 500 microbial species. Some approaches have also been undertaken to use machine learning models on NGS data to identify or classify reads that belong to novel emergent viruses. Zhang et al. [
20] as well as Bartoszewicz et al. [
21] employ diverse machine learning models to identify viruses that can infect humans. Whilst Zhang et al. applied the k-nearest neighbour (k-NN) model using
k-mer frequencies as input features for their classification, Bartoszewicz et al. employed a convolutional neural network (CNN) on simulated reads and contigs.
In this work, in contrast to the approaches by Zhang et al. and Bartoszewicz et al., the ability of an ANN to classify sequencing reads on the taxonomic level of nine biological orders was studied. Although the main focus of this work is on ANN models, we include linear discriminant analysis (LDA) and support vector machines (SVM) for comparison. The level of biological order was chosen since the data base is best suited among the different taxonomic levels, while the ANN studied by Rasheed et al. was fitted on sequence reads from a rather small number of microbes. We trained our model on the basis of several thousand viral sequences available at the NCBI database [
22]. In comparison to SVMs or LDA, ANNs are reported to achieve the highest accuracy rates on test data, although it depends on the specific classification problem [
23,
24]. However, despite being trained on a weak data base, an ANN can still produce a large number of misclassifications. Therefore, we additionally incorporated auxiliary test sets of sequencing reads with known taxonomic memberships and usde these to estimate conditional class probabilities in the individual taxonomic orders. These estimates were then used to correct estimated taxonomic distributions in the actual test sample. For that correction, we present new formulas to derive posterior class distributions.
Before fitting ANNs, we linked available viral reference sequences with their taxonomic description and studied the distribution of different taxonomic levels (phyla, subphyla, classes, subclasses, orders, etc.) to identify which of these ranks a solid data basis for building a training dataset would be reasonable. Next, we extracted k-mer distributions and inter-nucleotide distances as input features from the available reference sequences. In total, we generated 120 input features, and studied which combination qualified best for fitting an ANN.
Feature selection, building of the training data, the structure of the ANN model as well as the correction methods are provided in the following methods section. In the subsequent results section, a simulation study in which we cross-validate the performance of the ANN and the estimation of the taxonomic frequency distributions is presented. In addition, we compare the precision of the estimations with frequency distributions derived from a mapping approach. Finally, we discuss benefits and limitations of our approach.
2. Materials and Methods
2.1. Linking of Taxonomy and Genome Data
The taxonomy database containing data needed for taxonomic classification was downloaded from the ftp-server (
ftp://ftp.ncbi.nih.gov/refseq/release/viral, accessed on 15 March 2020) of the National Center for Biotechnology Information [
25] in March 2020. An extract of the taxonomy database file fullnamelineage.dmp from this ftp-server is depicted in
Figure 1. In this file, taxonomic information about various species are listed in rows. Beginning with archaea, bacteria and fungi, viral data is included from line 2,007,414 onwards. Row entries contain species names and taxonomy levels which are, however, neither labelled nor complete. We assigned these taxa to taxonomic levels based on the classification scheme of the International Committee on Taxonomy of Viruses [
26]. To enable correct assignments, we used the common taxa endings as specified by the International Committee on Taxonomy of Viruses [
26]. e.g., the ending ‘virus’ indicates the realm, the ending ‘vira’ the subrealm, the ending ‘virae’ the kingdom, etc. In
Figure 1, for example, the Bovine adenovirus 4 on line 2,007,418 is a member of the family of Adenoviruses.
As the taxonomic database does not provide any unique NC identifier, the species names of the reference genome file were searched in the taxonomy file to link genomic with taxonomic data. We obtained viral reference genomes as FASTA-files [
22] again from the NCBI database and concatenate the three files into a single one. By combining genomic and taxonomic data, we created a structured taxonomy table including FASTA-files of species which replaces the unstructured original taxonomy file. As the original files contain missing data, we only chose species with available taxonomy information. From more than 2 million taxonomy entries, only about 200,000 entries remain that are related to viruses. In contrast, the FASTA-file contains only 12,180 viral genome sequences and 2332 sequences provide taxonomy data. Hence, a maximum of 2332 viruses can be used in the training data.
2.2. Selection of a Suitable Taxonomic Category
From a list of 14 taxonomy levels (
Table 1), one needs to decide which one constitutes a suitable basis as a training set. As a necessary criterion, any classification algorithm requires at least two different taxonomy groups. Furthermore, to achieve a high accuracy, artificial neural networks need a large amount of training data in relation to the number of different groups one wants to predict. As a consequence, we looked for a taxonomic category that has more than one, but not too many different levels and that provides a sufficient amount of available viruses per level. e.g., the two taxonomic levels ‘family’ and ‘genus’ have a large number of annotated viruses, but too many different groups, each. The level ‘suborder’ would have a comfortable number of 5 groups, but too few annotated viruses. This is why we chose the taxonomy level of ‘orders’. Viral orders and the numbers of stored species per order in the NCBI database are
Bunyavirales (7),
Caudovirales (1859),
Herpesvirales (22),
Ligamenvirales (21),
Mononegavirales (70),
Nidovirales (47),
Ortervirales (81),
Picornavirales (107) and
Tymovirales (118). Take note, that the distribution is very unbalanced between the different order groups. For example, there are only seven viruses in the order
Bunyavirales, but 1859 viruses in the order
Caudovirales.
2.3. Sampling of Viral Reads for Training and Test Sets
Next, we detail how viruses and reads were sampled from which the input features are computed subsequently. We first used the FASTA-file provided by the NCBI database, restricted to 2332 viral genome sequences whose taxonomy data are available. From each order
, we randomly sampled the corresponding viruses without replacement and divided them randomly into training and test viruses. For optimizing the weights in an ANN, the training data was typically split into training and validation set. The disjunct partition of training, validation and test data was chosen as
,
and
, respectively. For example, from seven
Bunyavirales viruses, five were used for training, one for validation and one for the test data (
Table 2).
In step two, we randomly sampled with replacement the same amount of viruses out of each of the
cells in
Table 2. I.e., from each order
and for training, validation and test data separately (
), we sampled
of
viruses which yielded a total amount of
n viruses:
The whole sampling plan from step one and two is also known as two-stage stratified sampling with disproportionate allocation [
27]. After sampling the viruses, we sampled reads of the FASTA file by choosing random start positions out of a discrete uniform distribution over each viral genome and a read length of 150, a typical reads lengths in NGS.
2.4. Specification and Selection of ANN Input Features from Sequencing Reads
Possible input features to select for an ANN are for example
k-mers [
28], inter-nucleotide distances [
29] and DNA sequence motifs [
30]. A
k-mer is a subsequence of length
k of the original read sequence. Inter-nucleotide distances are the distances between two identical bases. For instance, the DNA sequence TTATACTACGTGGGGGGGGGTCCT exhibits T-T distances of 1, 2, 3, 4, 10 and 3 and we are interested in the count frequencies of the distances 2 to 10. A DNA sequence motif is a set of words which can have arbitrary or a subset of A, C, G and T letters on particular base positions while having fixed letters on other positions. Here, we focus on DNA sequence motifs with two or three fixed letters, therefrom two at the margins, such as C.....T or G..G...A.
The overall number of input features studied in our analysis, sums up to where are the numbers of k-mers, 36 inter-nucleotide distances and and the numbers of selected DNA sequence motifs with two or three fixed letters, respectively. As we will describe below, no DNA sequence motifs were selected. Therefore, 120 input features were finally studied. We did not include 4-mers or DNA sequence motifs with four fixed letters because this would result in too many input features and the probability of obtaining frequencies equal zero was too high.
Selection of input features was based on the results of Kruskal–Wallis tests and by accuracy comparisons in additional ANN simulations. Separately for each sequence motif, the relative frequency in each complete nucleobase sequence of the FASTA-file was counted. Next, the frequencies between and within the groups or orders were compared by Kruskal–Wallis tests [
31]. High differences of the frequencies between the orders in relation to low differences within the orders point out that this particular DNA sequence motif would be a good input feature to differentiate between the orders. Finally, we selected those sequence motifs with the lowest
p-value and highest effect size
[
32], respectively, up to a sequence length of eight. From all possible DNA sequence motifs with two or three fixed letters, we chose those with an effect size not less than
or
, respectively.
We iterated 10 simulation runs per layer in the artificial neural network and evaluated mean and standard deviation of accuracy results (see
Table 3). Each layer uses a subset of all proposed input features. If the paired
t-test recognises a statistically significant and relevant accuracy increase of the subsequent layer, its corresponding input features are added to the ANN model. As a results, the feature selection method advocates to use 1-mers, 2-mers, 3-mers and inter-nucleotide distances as input features.
2.5. Artificial Neural Net Structure
For computation of the ANN [
33], we applied the R-package keras [
34]. The sequential model we used contains 100,000 training reads per virus order. The input layer contains 120 input nodes, the hidden layer 64 nodes and the output layer nine nodes representing the nine different orders. The activation functions used were ‘Rectified Linear Unit’ (ReLU) [
35] for the hidden layer and ‘Softmax’ [
36] for multinomial classification of the output layer. As an optimisation algorithm we chose ‘stochastic gradient descent’. To quantify the model performance, sparse categorical crossentropy and accuracy were used. In order to study whether a more complex ANN can reach more accuracy, we also included a model with 5 hidden layers, each again with 64 nodes and the ReLU activation function.
While training the model, the data were shuffled before each epoch to allow for faster convergence and to prevent memorizing of the order of the training samples [
37]. Batch sizes of
were used as well as a maximum number of training epochs of 500 as an early stopping method to prevent overfitting [
38].
2.6. Building of the Confusion Matrix and Measures of Classification Performance
The typical summary table of classification results is a confusion matrix. In our case, cell entry of the confusion matrix provided the number of reads of true order j classified to order k (). In our particular test data sets, 21,429 reads per order were given, represented by the row-sums of the confusion matrix. The diagonal from top left to bottom right provides the correct classification frequencies from which the accuracy can be derived.
As additional performance metrics, the true positive rate (TPR, sensitivity) and true negative rate (TNR, specificity), as well as positive and negative predictive values (PPV and NPV) can be reported. These four measures must be interpreted in the sense of one biological order versus the eight other orders, here.
2.7. Estimation Correction of Taxa Frequencies
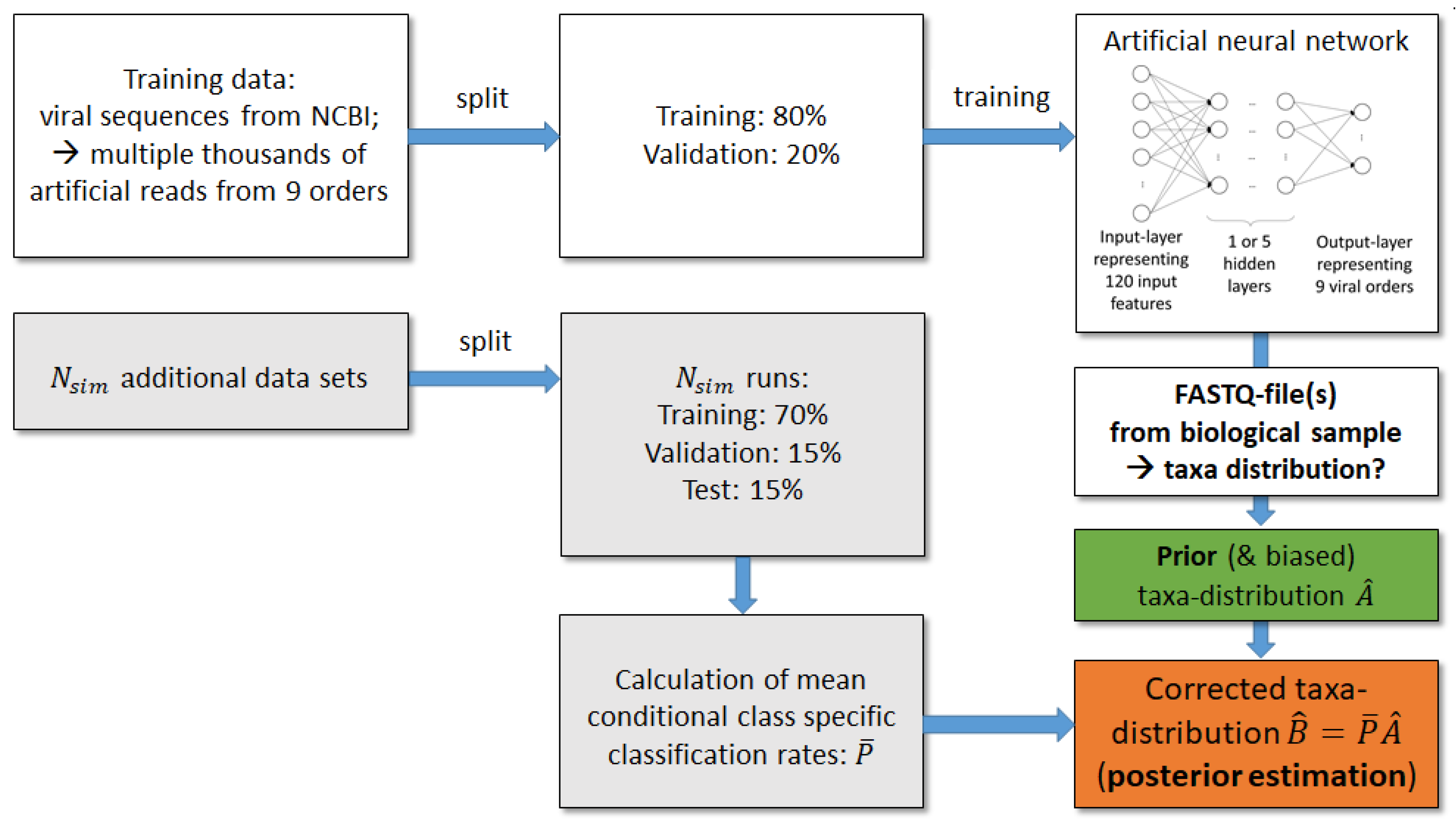
The artificial neural network classifies single reads into taxonomic orders. However, we do not only want to predict to which order a particular read belongs, but also want to estimate the global taxa frequency distribution of the nine orders in a biological sample. In this way we gain a summary of species abundance on taxonomic levels. The number of reads assigned by the ANN to the nine orders provides a prior frequency distribution. Since the sequences of viruses within one order can have little similarity, a good classification performance of an ANN is still a challenge, and a large number of misclassifications and thus a biased prior estimation of taxa distributions is likely. Therefore, we employed additional auxiliary test sets of labelled reads to determine conditional probabilities that a read classified to a particular order belongs to this one or to one of the other orders. We then used this additional information to estimate a posterior, less unbiased distribution. The correction procedure is described in the following (cf.
Figure 2).
We denoted the number of auxiliary data sets as
and the number of taxonomic categories as
; in our scenario the categories are the 9 viral orders. Each of the
auxiliary data sets consists of a training and a test sample. The training sample was further split into training and validation set to fit the parameters of the ANN. The fitted ANN was then applied to the auxiliary test sets resulting in
confusion matrices:
with true order labels in rows and predicted labels in columns. The column sums from the matrices
compose a prior estimation
of the read count distribution in the nine orders. Furthermore, the confusion matrices
were used to calculate the matrices
which contain the conditional class probabilities that given a read classified to order
, thus it truly belongs to order
. From the matrices
leave-one-out mean matrices
can be calculated using all matrices
except the one corresponding to the particular simulation run.
By applying the law of the total probability and multiplying each leave-one-out mean matrix
by the related
s-th column of
, we receive the posterior estimations of read count distributions for the auxiliary test samples
s:
i.e., in test run
s the posterior probability that a read in the sample belongs to order
j is given by the sum of conditional probabilities of order
j multiplied by the prior probabilities of orders
k in run
s:
Thus, the read count distribution of run s is corrected by information about the conditional class probabilities estimated from the independent other simulation runs.
When analysing a real-world test sample with unknown class memberships, matrices would be obtained from the above simulations with auxiliary test sets and the classification of sequencing reads from the real world sample would result in a new matrix with dimension . Since test samples were generated artificially, the corrected (i.e., posterior) distribution of taxa would instead have dimension .
3. Results
Our primary research question was to study the ability of an ANN to classify sequencing reads with unknown taxonomic order and to subsequently deriving the frequency distribution of these orders in the sample. First, we present the achieved accuracy of our fitted models, and demonstrate then how our correction approach can be used to obtain an improved estimation of the taxonomic order frequency distribution. The validation of our approach was performed on simulated sequencing data with runs. In each run, different sets of viruses and viral reads were drawn. Finally, we demonstrate the effect of the correction approach in a publicly available sequencing sample taken from a harbour seal.
3.1. Performance History of the Network Fitting Processes and Results from Other Machine Learning Models
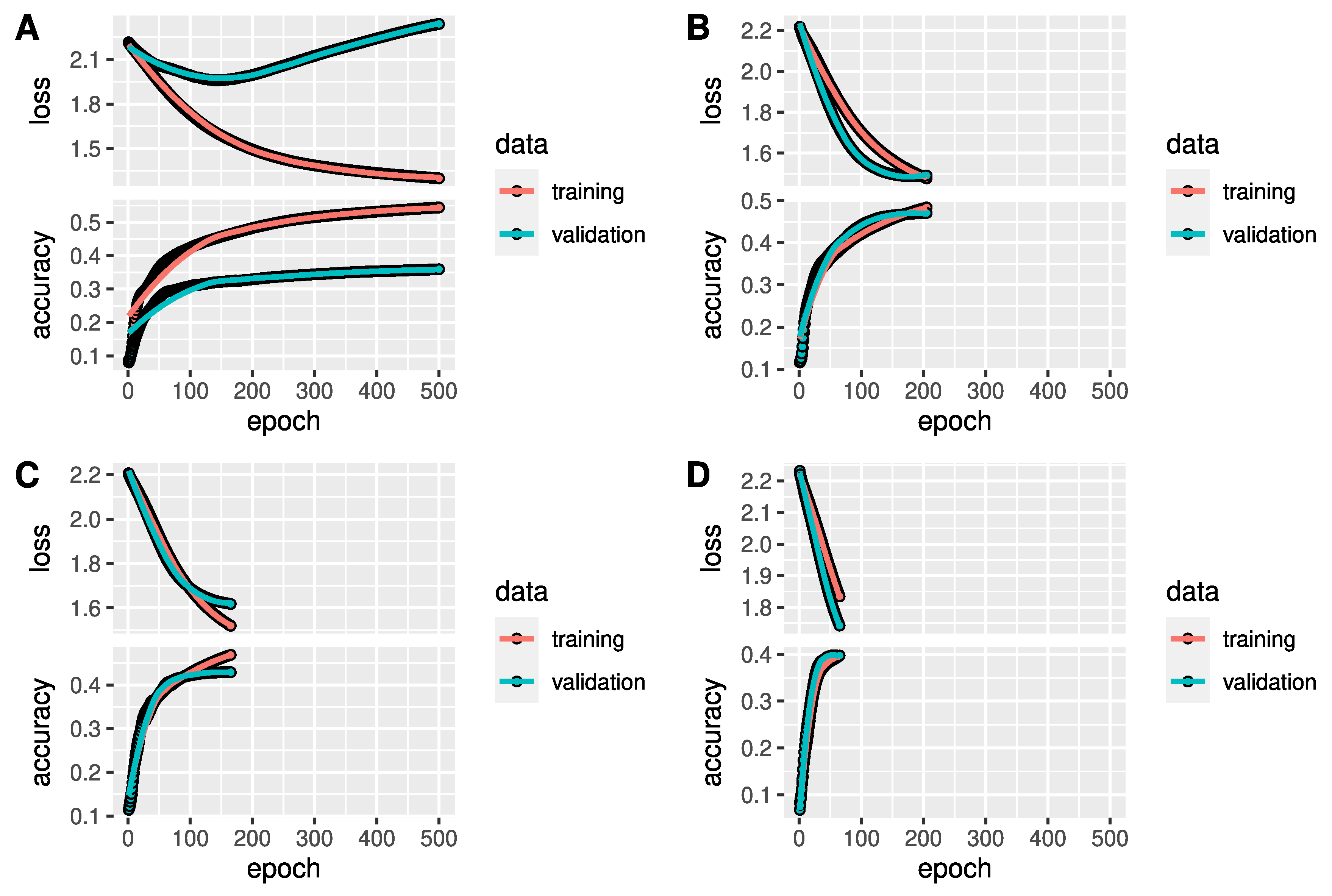
Exemplarily, the performance history of 4 out of 10 simulation runs is depicted in
Figure 3 in form of the loss and accuracy with respect to the training epoch of the ANNs with on hidden layer. Accuracy and loss curves are presented for training and validation data. We allowed the maximal number of epochs to be 500, however, in some runs, the fitting converged much faster. Training loss decreased negatively exponential and training accuracy increased logarithmically. After the final number of epochs was reached, validation accuracy was considerably higher than
in all ten runs. Considering that we have nine taxonomic orders, this means that the ANN classifications were at least more precise than random classifications. Plot A shows an exceptional validation performance behaviour. Validation loss decreased slightly until epoch 150, then increased to a value higher than at the start, while validation accuracy was still increasing. This indicates that the uncertainty for predicting class probabilities increases, whereas the classification results are becoming more precise. Plots B to D partly show better loss and accuracy performances on validation data in comparison to training data.
The confusion matrix in
Table 4 shows true (rows) versus classified (columns) orders. Presented counts are averaged over the 10 simulation runs. For each order
j, classification results were additionally treated as results from a binary classifier, i.e., order
j versus all other orders. Accordingly, sensitivity (i.e., true positive rate, TPR), specificity (true negative rate, TNR), positive (PPV) and negative predictive values (NPV) were derived. Averages of these four measures over all 10 simulation runs are shown in
Table 5, while TNRs and NPVs are high for all orders, TPRs and PPV are rather weak.
Although achieved accuracies and the other measures of performance were better than by a random classifier, the fitted ANNs still resulted in a large number of misclassifications. Consequently, the estimated frequency distributions for the nine biological orders are biased. These results were for us the reason to develop the correction procedure detailed in the methods section. Simulated findings for this correction procedure are provided in the next subsection.
The mean accuracy of the simple ANN model to correctly classify a read as derived from the 10 simulation runs was 0.40 (minimum: 0.35, maximum: 0.46.), which is higher than a random classifier and would result in an accuracy of 0.11, due to the nine order. We also checked whether a more complex ANN with five hidden layers, or other machine learning approaches, explicitly LDA or SVMs with different kernels, can improve the accuracy, while the ANN with five hidden layers and the SVM with polynomial kernel achieved nearly the same (but also no better) accuracy than the ANN with only one layer, the other models achieved clearly worse accuracies (
Table 6).
3.2. Prior and Corrected Estimation of Taxa Frequencies
Boxplots in
Figure 4A show the distributions of mapping, prior and posterior (i.e., corrected) estimation of taxa frequencies of the nine biological orders obtained in 10 simulation runs for a true balanced taxa distribution, i.e., with 1/9∼11% of reads per order in training, validation and test set, and when using an ANN with one hidden layer. Both, variance and bias, and thus mean squared error of posterior estimation, are considerably smaller for each order in comparison to prior or mapping estimation. As was seen with the accuracy in the previous section, no clear improvement of the posterior estimation can be observed when fitting a more complex ANN model with five hidden layers (
Figure 4B).
Furthermore, frequency estimation by the mapping approach performed worst among the competing approaches. In general, the mapping rates were low with a mean value of and a standard deviation of which can be traced back to the fact the viral reads in the test data were disjunct to those in the training data. Among the mapped reads, not a single one was mapped to a virus from the orders Bunyavirales or Tymovirales, and consequently taxa frequencies of these orders were underestimated. In contrast, Caudovirales and Ligamenvirales orders were overestimated considerably. On the median level, more than of all reads were mapped to Caudovirales although only were expected. Since the difference in the statistical estimation error of the mapping approach in comparison to the ANN approaches is graphically obvious, no statistical test was performed to proof its inferiority.
Besides the graphical comparison, we applied Mood’s median test to compare the median absolute deviation of estimated frequencies from the true value of 21,429 (i.e., ) test reads between the prior and posterior estimation. The resulting p-value is < which is significant when taking an of . Median absolute deviations of prior and posterior estimation were or reads, respectively, which translates to a difference of reads. Thus, median posterior estimation of taxa frequencies is over 6000 reads more precise than median prior estimation.
In order to see whether the correction approach also works for other machine learning approaches than ANNs, we applied it to the SVM with polynomial kernel and LDA, while there are clearly different prior estimations obtained from SVM and LDA, the posterior estimation again is able to correct this bias from the prior estimations (
Figure 5). Corresponding to the low accuracy for read classification, the bias of the prior taxa estimation with LDA is also much worse than that of the SVM and the ANN.
While the above scenarios where based on the same distributions of viral orders in the training, validation and test sets, we also simulated more realistic scenarios with different taxa distributions in these data sets. In particular, the distributions for training and validation set were kept balanced (i.e., 11% of reads per set), while the distributions of the test sets where drawn randomly from the Dirichlet-multinomial distribution [
39]. In a real-world scenario, the analyst would not be restricted to use other than balanced distribution for training and validation set; however, a true test set from a biological sample would most likely show an unbalanced distribution of the nine orders. We simulated two of such example scenarios and fitted again ANNs with one hidden layer. In both scenarios, there is a clear bias by the prior estimation, while the posterior estimation corrects this bias well (
Figure 6).
3.3. Effect of Frequency Correction in Sequencing Data from a Harbour Seal
To demonstrate how biased and corrected frequency estimation can affect the biological conclusion in a real sample, we applied the machine learning approach on a FASTQ-file generated from a biological sample of a harbour seal [
40]. In this real-world sample, we do not know the true distribution of orders, thus this example can only demonstrate the shift between prior and posterior estimation. After mapping to a database of viral reference genomes [
22] and filtering of low quality reads with a mapping quality smaller than 2, 3,154,562 sequencing reads remained for classification and estimation of taxa frequencies. Following the procedure depicted in
Figure 2, we first received prior estimations of order frequencies, and used
auxiliary data sets to determine conditional classification probabilities
. The latter information was used to correct prior estimations according to formula (1). In contrast to the above simulation, only one frequency value per biological order is obtained for the prior estimation (
Figure 7). Due to the 10 auxiliary data sets posterior results provide a frequency range for each order, while the prior estimation yields nearly zero percent frequency for 5 of the studied orders, correction shows a slightly changed distribution, which could have a strong impact on the biological interpretation of a sample within a metagenomics analysis.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}