
A Comprehensive, Targeted NGS Approach to Assessing Molecular Diagnosis of Lysosomal Storage Diseases



Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Dosage
2.2. Panel Design and Library Preparation
2.3. Chip Loading and Sequencing
2.4. Variant Calling and Prioritization
3. Results
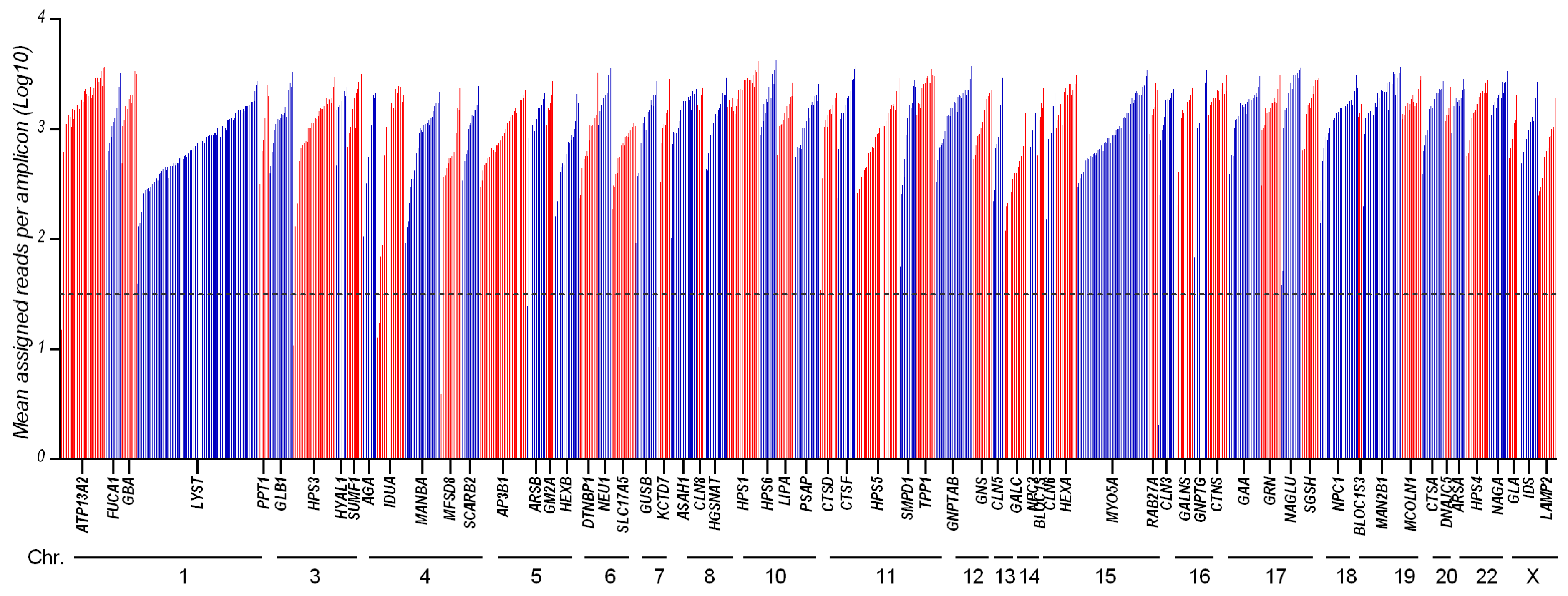
3.1. Panel Design and Performance
3.2. Control Samples Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Platt, F.M. Emptying the stores: Lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 2018, 17, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Gahl, W.A. Lysosomal storage diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71. [Google Scholar] [CrossRef] [Green Version]
- La Cognata, V.; Guarnaccia, M.; Polizzi, A.; Ruggieri, M.; Cavallaro, S. Highlights on Genomics Applications for Lysosomal Storage Diseases. Cells 2020, 9, 1902. [Google Scholar] [CrossRef]
- Kingma, S.D.; Bodamer, O.A.; Wijburg, F.A. Epidemiology and diagnosis of lysosomal storage disorders; challenges of screening. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 145–157. [Google Scholar] [CrossRef]
- Pinto, E.V.F.; Rojas Malaga, D.; Kubaski, F.; Fischinger Moura de Souza, C.; de Oliveira Poswar, F.; Baldo, G.; Giugliani, R. Precision Medicine for Lysosomal Disorders. Biomolecules 2020, 10, 1110. [Google Scholar] [CrossRef] [PubMed]
- Mokhtariye, A.; Hagh-Nazari, L.; Varasteh, A.R.; Keyfi, F. Diagnostic methods for Lysosomal Storage Disease. Rep. Biochem. Mol. Biol. 2019, 7, 119–128. [Google Scholar]
- Waggoner, D.J.; Tan, C.A. Expanding newborn screening for lysosomal disorders: Opportunities and challenges. Dev. Disabil. Res. Rev. 2011, 17, 9–14. [Google Scholar] [CrossRef]
- Komlosi, K.; Sólyom, A.; Beck, M. The Role of Next-Generation Sequencing in the Diagnosis of Lysosomal Storage Disorders. J. Inborn Errors Metab. Screen. 2016, 4, 4. [Google Scholar] [CrossRef]
- Encarnação, M.; Coutinho, M.F.; Silva, L.; Ribeiro, D.; Ouesleti, S.; Campos, T.; Santos, H.; Martins, E.; Cardoso, M.T.; Vilarinho, L.; et al. Assessing Lysosomal Disorders in the NGS Era: Identification of Novel Rare Variants. Int. J. Mol. Sci. 2020, 21, 6355. [Google Scholar] [CrossRef]
- Fernandez-Marmiesse, A.; Morey, M.; Pineda, M.; Eiris, J.; Couce, M.L.; Castro-Gago, M.; Fraga, J.M.; Lacerda, L.; Gouveia, S.; Perez-Poyato, M.S.; et al. Assessment of a targeted resequencing assay as a support tool in the diagnosis of lysosomal storage disorders. Orphanet J. Rare Dis. 2014, 9, 59. [Google Scholar] [CrossRef]
- Zanetti, A.; D’Avanzo, F.; Bertoldi, L.; Zampieri, G.; Feltrin, E.; De Pascale, F.; Rampazzo, A.; Forzan, M.; Valle, G.; Tomanin, R. Setup and Validation of a Targeted Next-Generation Sequencing Approach for the Diagnosis of Lysosomal Storage Disorders. J. Mol. Diagn. 2020, 22, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Gheldof, A.; Seneca, S.; Stouffs, K.; Lissens, W.; Jansen, A.; Laeremans, H.; Verloo, P.; Schoonjans, A.S.; Meuwissen, M.; Barca, D.; et al. Clinical implementation of gene panel testing for lysosomal storage diseases. Mol. Genet. Genom. Med. 2019, 7, e00527. [Google Scholar] [CrossRef]
- Levesque, S.; Auray-Blais, C.; Gravel, E.; Boutin, M.; Dempsey-Nunez, L.; Jacques, P.E.; Chenier, S.; Larue, S.; Rioux, M.F.; Al-Hertani, W.; et al. Diagnosis of late-onset Pompe disease and other muscle disorders by next-generation sequencing. Orphanet J. Rare Dis. 2016, 11, 8. [Google Scholar] [CrossRef]
- Di Fruscio, G.; Schulz, A.; De Cegli, R.; Savarese, M.; Mutarelli, M.; Parenti, G.; Banfi, S.; Braulke, T.; Nigro, V.; Ballabio, A. Lysoplex: An efficient toolkit to detect DNA sequence variations in the autophagy-lysosomal pathway. Autophagy 2015, 11, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Malaga, D.R.; Brusius-Facchin, A.C.; Siebert, M.; Pasqualim, G.; Saraiva-Pereira, M.L.; Souza, C.F.M.; Schwartz, I.V.D.; Matte, U.; Giugliani, R. Sensitivity, advantages, limitations, and clinical utility of targeted next-generation sequencing panels for the diagnosis of selected lysosomal storage disorders. Genet. Mol. Biol. 2019, 42, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Cognata, V.; Guarnaccia, M.; Morello, G.; Ruggieri, M.; Polizzi, A.; Cavallaro, S. Design and Validation of a Custom NGS Panel Targeting a Set of Lysosomal Storage Diseases Candidate for NBS Applications. Int. J. Mol. Sci. 2021, 22, 64. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, M.M.; Cuthbert, C.D.; Cordovado, S.K. Assessing the Performance of Dried-Blood-Spot DNA Extraction Methods in Next Generation Sequencing. Int. J. Neonatal Screen. 2020, 6, 36. [Google Scholar] [CrossRef]
- Boemer, F.; Fasquelle, C.; d’Otreppe, S.; Josse, C.; Dideberg, V.; Segers, K.; Guissard, V.; Capraro, V.; Debray, F.G.; Bours, V. A next-generation newborn screening pilot study: NGS on dried blood spots detects causal mutations in patients with inherited metabolic diseases. Sci. Rep. 2017, 7, 17641. [Google Scholar] [CrossRef]
- Davidson, B.A.; Hassan, S.; Garcia, E.J.; Tayebi, N.; Sidransky, E. Exploring genetic modifiers of Gaucher disease: The next horizon. Hum. Mutat. 2018, 39, 1739–1751. [Google Scholar] [CrossRef]
- Parenti, G.; Medina, D.L.; Ballabio, A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol. Med. 2021, 13, e12836. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Gene | Cytogenetic Location | Pathology | Phenotype OMIM No. |
|---|---|---|---|
| AGA | 4q34.3 | Aspartylglucosaminuria | 208400 |
| AP3B1 | 5q14.1 | Hermansky–Pudlak disease type 2 | 608233 |
| ARSA | 22q13.33 | Metachromatic leukodystrophy | 250100 |
| ARSB | 5q14.1 | MPS VI, also known as Maroteaux–Lamy syndrome | 253200 |
| ASAH1 | 8p22 | Farber lipogranulomatosis | 228000 |
| ATP13A2 | 1p36.13 | CLN12b: Kufor–Rakeb syndrome or PARK9 | 606693 |
| BLOC1S6 | 15q21.1 | Hermansky–Pudlak disease type 9 | 614171 |
| BLOCS13 | 19q13.32 | Hermansky–Pudlak disease type 8 | 614077 |
| CLN3 | 16p12.1 | CLN3: Batten–Spielmeyer–Sjogren disease | 204200 |
| CLN5 | 13q22.3 | CLN5: Finnish variant late infantile | 256731 |
| CLN6 | 15q23 | CLN6: Lake–Cavanagh or Indian variant | 601780 |
| CLN8 | 8p23.3 | CLN8: northern epilepsy, epilepsy mental retardation | 600143 610003 |
| CTNS | 17p13.2 | Cystinosis | 219800 |
| CTSA | 20q13.12 | Galactosialidosis | 256540 |
| CTSD | 11p15.5 | CLN10 | 610127 |
| CTSF | 11q13.2 | CLN13 | 615362 |
| DNAJC5 | 20q13.33 | CLN4: Parry disease and Kufs type A and B | 162350 |
| DTNBP1 | 6p22.3 | Hermansky–Pudlak disease type 7 | 614076 |
| FUCA1 | 1p36.11 | Fucosidosis | 230000 |
| GAA | 17q25.3 | Pompe disease | 232300 |
| GALC | 14q31.3 | Globoid cell leukodystrophy, Krabbe disease | 245200 |
| GALNS | 16q24.3 | MPS IVA, also known as Morquio syndrome A | 253000 |
| GBA | 1q22 | Gaucher disease | 230800 |
| GLA | Xq22.1 | Fabry disease | 301500 |
| GLB1 | 3p22.3 | GM1 gangliosidosis; MPS IVB, also known as Morquio syndrome B | 253010 |
| GM2A | 5q33.1 | GM2 gangliosidosis, GM2 activator deficiency | 272750 |
| GNPTAB | 12q23.2 | Mucolipidosis II α/β, I-cell disease; mucolipidosis III α/β, pseudo-Hurler polydystrophy | 252500 252600 |
| GNPTG | 16p13.3 | Mucolipidosis III γ, variant pseudo-Hurler polydystrophy | 252605 |
| GNS | 12q14.3 | MPS IIID, also known as Sanfilippo syndrome D | 252940 |
| GRN | 17q21.31 | CLN11 | 614706 |
| GUSB | 7q11.21 | MPS VII, also known as Sly disease | 253220 |
| HEXA | 15q23 | GM2 gangliosidosis, Tay–Sachs disease | 272800 |
| HEXB | 5q13.3 | GM2 gangliosidosis, Sandhoff diseaseb | 268800 |
| HGSNAT | 8p11.2-p11.1 | MPS IIIC, also known as Sanfilippo syndrome C | 252930 |
| HPS1 | 10q24.2 | Hermansky–Pudlak disease type 1 | 203300 |
| HPS3 | 3q24 | Hermansky–Pudlak disease type 3 | 614072 |
| HPS4 | 22q12.1 | Hermansky–Pudlak disease type 4 | 614073 |
| HPS5 | 11p15.1 | Hermansky–Pudlak disease type 5 | 614074 |
| HPS6 | 10q24.32 | Hermansky–Pudlak disease type 6 | 614075 |
| HYAL1 | 3p21.31 | MPS IX | 601492 |
| IDS | Xq28 | MPS II, also known as Hunter syndrome | 309900 |
| IDUA | 4p16.3 | MPS I: Hurler syndrome | 607014 607015 607016 |
| KCTD7 | 7q11.21 | CLN14 | 611726 |
| LAMP2 | Xq24 | Danon disease | 300257 |
| LIPA | 10q23.31 | Acid lipase deficiency: Wolman disease and cholesterol ester storage disease | 278000 |
| LYST | 1q42.3 | Chédiak–Higashi disease | 214500 |
| MAN2B1 | 19p13.13 | α-Mannosidosis | 248500 |
| MANBA | 4q24 | β-Mannosidosis | 248510 |
| MCOLN1 | 19p13.2 | Mucolipidosis IV | 252650 |
| MFSD8 | 4q28.2 | CLN7: Turkish variant | 610951 |
| MYO5A | 15q21.2 | Griscelli syndrome 1, also known as Elejalde syndrome | 214450 |
| NAGA | 22q13.2 | Schindler disease: type Ib, also known as infantile-onset neuroaxonal dystrophy, type IIb also known as Kanzaki disease, and type IIIb, intermediate severity | 609241 609242 |
| NAGLU | 17q21.2 | MPS IIIB, also known as Sanfilippo syndrome B | 252920 |
| NEU1 | 6p21.33 | Sialidosis type I, Sialidosis type II | 256550 |
| NPC1 | 18q11.2 | Niemann–Pick disease types C1 | 257220 |
| NPC2 | 14q24.3 | Niemann–Pick disease types C1 and C2 | 607625 |
| PPT1 | 1p34.2 | CLN1: Haltia–Santavuori disease and INCL | 256730 |
| PSAP | 10q22.1 | Metachromatic leukodystrophy | 249900 |
| RAB27A | 15q21.3 | Griscelli syndrome 2 | 607624 |
| SCARB2 | 4q21.1 | Action myoclonus-renal failure syndrome | 254900 |
| SGSH | 17q25.3 | MPS IIIA, also known as Sanfilippo syndrome A | 252900 |
| SLC17A5 | 6q13 | Sialic acid storage disease | 269920 |
| SMPD1 | 11p15.4 | Niemann–Pick disease types A and B | 257200 607616 |
| SUMF1 | 3p26.1 | Multiple sulfatase deficiency | 272200 |
| TPP1 | 11p15.4 | CLN2, also known as Jansky–Bielschowsky disease | 204500 |
| ID Coriell Sample | Genes | Zigosity | Transcript | Coding Amino Acid Change | Variant Effect | dbSNP | ClinVar |
|---|---|---|---|---|---|---|---|
| NA03392 | GNPTG | Hom | NM_032520.5 | c.445delG p.Ala149ProfsTer13 | frameshiftDeletion | rs1555451874 | P |
| NA03461 | HEXA | Het | NM_000520.6 | c.1421+1G>C p.? | unknown | rs147324677 | P |
| c.805G>A p.Gly269Ser | missense | rs121907954 | P/LP | ||||
| NA05093 | GNS | Hom | NM_002076.4 | c.1063C>T p.Arg355Ter | nonsense | rs119461974 | P |
| NA00654 | GLB1 | Het | NM_000404.4 | c.1032T>C p.Thr344= | synonymous | rs199927127 | CIP |
| MAN2B1 | Het | NM_000528.4 | c.2248C>T p.Arg750Trp | missense | rs80338680 | P | |
| c.1915C>T p.Gln639Ter | nonsense | rs121434332 | P | ||||
| NA02528 | AP3B1 | Het | NM_003664.5 | c.1168-9C>T p.? | unknown | rs367648410 | CIP |
| MCOLN1 | Hom | NM_020533.3 | c.406-2A>G p.? | unknown | rs104886461 | P | |
| NA01675 | MFSD8 | Het | NM_152778.3 | c.590G>A p.Gly197Asp | missense | rs28544073 | CIP |
| GM2A | Hom | NM_000405.5 | c.412T>C p.Cys138Arg | missense | rs137852797 | P | |
| NA02455 | GLB1 | Het | NM_000404.4 | c.1445G>A p.Arg482His | missense | rs72555391 | P |
| c.817_818delTGinsCT p.Trp273Leu | missense | rs1559401428 | P/LP | ||||
| CLN6 | Het | NM_017882.3 | c.821C>T p.Ala274Val | missense | rs202012876 | US | |
| NA02013 | GNPTAB | Het | NM_024312.5 | c.3501_3502delTC p.Leu1168GlnfsTer5 | frameshiftDeletion | rs34002892 | P |
| c.3233_3234insCCTA p.Tyr1079LeufsTer3 | frameshiftInsertion | - | n.a. | ||||
| GNPTG | Het | NM_032520.5 | c.574G>C p.Glu192Gln | missense | rs749314645 | US | |
| NA02552 | GLB1 | Het | NM_000404.4 | c.602G>A p.Arg201His | missense | rs189115557 | P |
| HPS1 | Het | NM_000195.5 | c.29G>T p.Gly10Val | missense | rs759539605 | n.a. | |
| NAGLU | Het | NM_000263.4 | c.889C>T p.Arg297Ter | nonsense | rs104894592 | P/LP | |
| c.1928G>A p.Arg643His | missense | rs104894593 | US | ||||
| NA17881 | HPS6 | Hom | NM_024747.6 | c.1714_1717delCTGT p.Leu572AlafsTer40 | frameshiftDeletion | rs281865113 | P |
| NA17890 | LYST | Het | NM_000081.4 | c.149G>A p.Arg50Gln | missense | rs368095341 | n.a. |
| AP3B1 | Het | NM_003664.5 | c.1975G>T p.Glu659Ter | nonsense | rs121908907 | P | |
| c.1525C>T p.Arg509Ter | nonsense | rs121908906 | P | ||||
| NA17721 | SLC17A5 | Hom | NM_012434.5 | c.115C>T p.Arg39Cys | missense | rs80338794 | P |
| NA16081 | PPT1 | Het | NM_000310.4 | c.451C>T p.Arg151Ter | nonsense | rs137852700 | P/LP |
| c.236A>G p.Asp79Gly | missense | rs137852697 | P | ||||
| NA13204 | DTNBP1 | Het | NM_032122.5 | c.489_490insT p.Lys164Ter | nonsense | - | n.a. |
| HEXA | Het | NM_000520.6 | c.1277_1278insTATC p.Tyr427IlefsTer5 | frameshiftInsertion | rs387906309 | P | |
| c.805G>A p.Gly269Ser | missense | rs121907954 | P/LP | ||||
| NA18455 | MANBA | Het | NM_005908.4 | c.1442A>C p.Tyr481Ser | missense | rs764041854 | n.a. |
| NPC2 | Het | NM_006432.5 | c.140G>T p.Cys47Phe | missense | rs1555345993 | US | |
| c.58G>T p.Glu20Ter | nonsense | rs80358260 | P | ||||
| NA20387 | TPP1 | Het | NM_000391.4 | c.622C>T p.Arg208Ter | nonsense | rs119455955 | P |
| c.509-1G>C p.? | unknown | rs56144125 | P | ||||
| GALNS | Het | NM_000512.5 | c.858G>A p.Thr286= | synonymous | rs140299014 | CIP | |
| NA20019 | ASAH1 | Het | NM_004315.6 | c.1039G>A p.Asp347Asn | missense | rs1354060089 | US |
| c.460G>T p.Glu154Ter | nonsense | rs1588982399 | LP | ||||
| GNPTAB | Het | NM_024312.5 | c.2708_2710delTTC p.Leu904del | nonframeshiftDeletion | rs774128798 | US | |
| NA10866 | IDUA | Het | NM_000203.5 | c.785A>G p.His262Arg | missense | rs1031451164 | n.a. |
| IDS | Hom | NM_000202.8 | c.1403G>C p.Arg468Pro | missense | rs113993946 | P | |
| NA12928 | HPS1 | Hom | NM_000195.5 | c.1484_1485insCCCCCAGCAGGGGAGG p.His497GlnfsTer90 | frameshiftInsertion | - | n.a. |
| HPS6 | Het | NM_024747.6 | c.2250G>A p.Ser750= | synonymous | rs139161525 | CIP | |
| MYO5A | Het | NM_000259.3 | c.3567+4C>T p.? | unknown | rs186277072 | n.a. | |
| NA06110 | SGSH | Het | NM_000199.5 | c.734G>A p.Arg245His | missense | rs104894635 | P |
| Het | c.629G>A p.Trp210Ter | nonsense | rs886041370 | P/LP | |||
| NA20379 | PPT1 | Het | NM_000310.4 | c.364A>T p.Arg122Trp | missense | rs137852695 | P |
| c.125G>A p.Gly42Glu | missense | rs386833631 | LP | ||||
| GAA | Het | NM_001079804.3 | c.525delT p.Glu176ArgfsTer45 | frameshiftDeletion | rs386834235 | P | |
| NA03124 | GUSB | Het | NM_000181.4 | c.454G>A p.Asp152Asn | missense | rs149606212 | US |
| NPC1 | Het | NM_000271.5 | c.3182T>C p.Ile1061Thr | missense | rs80358259 | P | |
| c.1947+5G>C p.? | unknown | rs770321568 | CIP | ||||
| ARSA | Het | NM_001085425.3 | c.698_699insC p.Gln234SerfsTer41 | frameshiftInsertion | - | n.a. | |
| NA03111 | LIPA | Het | NM_001127605.3 | c.967_968delAG p.Ser323LeufsTer44 | frameshiftDeletion | rs917089035 | n.a. |
| c.894G>A p.Gln298= | synonymous | rs116928232 | P/LP | ||||
| GALNS | Het | NM_000512.5 | c.499T>G p.Phe167Val | missense | rs148565559 | US | |
| NA02057 | AGA | Het | NM_000027.4 | c.488G>C p.Cys163Ser | missense | rs121964904 | P |
| NA00879 | BLOC1S6 | Het | NM_012388.4 | c.225-2_225-1insT p.? | unknown | - | n.a. |
| SGSH | Het | NM_000199.5 | c.1339G>A p.Glu447Lys | missense | rs104894639 | P/LP | |
| SGSH | Second Variant not detected c.746G>A (Arg245His (R245H)) | ||||||
| CTSA | Het | NM_000308.4 | c.263_264insG p.Cys88TrpfsTer52 | frameshiftInsertion | - | n.a. | |
| NA01256 | IDUA | Het | NM_000203.5 | c.590-7G>A p.? | unknown | rs762411583 | P |
| Second Variant excluded because of very low coverage c.1293TGG>TAG (Trp402Ter (W402X)) | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Cognata, V.; Cavallaro, S. A Comprehensive, Targeted NGS Approach to Assessing Molecular Diagnosis of Lysosomal Storage Diseases. Genes 2021, 12, 1750. https://doi.org/10.3390/genes12111750
La Cognata V, Cavallaro S. A Comprehensive, Targeted NGS Approach to Assessing Molecular Diagnosis of Lysosomal Storage Diseases. Genes. 2021; 12(11):1750. https://doi.org/10.3390/genes12111750
Chicago/Turabian StyleLa Cognata, Valentina, and Sebastiano Cavallaro. 2021. "A Comprehensive, Targeted NGS Approach to Assessing Molecular Diagnosis of Lysosomal Storage Diseases" Genes 12, no. 11: 1750. https://doi.org/10.3390/genes12111750
APA StyleLa Cognata, V., & Cavallaro, S. (2021). A Comprehensive, Targeted NGS Approach to Assessing Molecular Diagnosis of Lysosomal Storage Diseases. Genes, 12(11), 1750. https://doi.org/10.3390/genes12111750