
Expanding the Phenotype of the FAM149B1-Related Ciliopathy and Identification of Three Neurogenetic Disorders in a Single Family

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
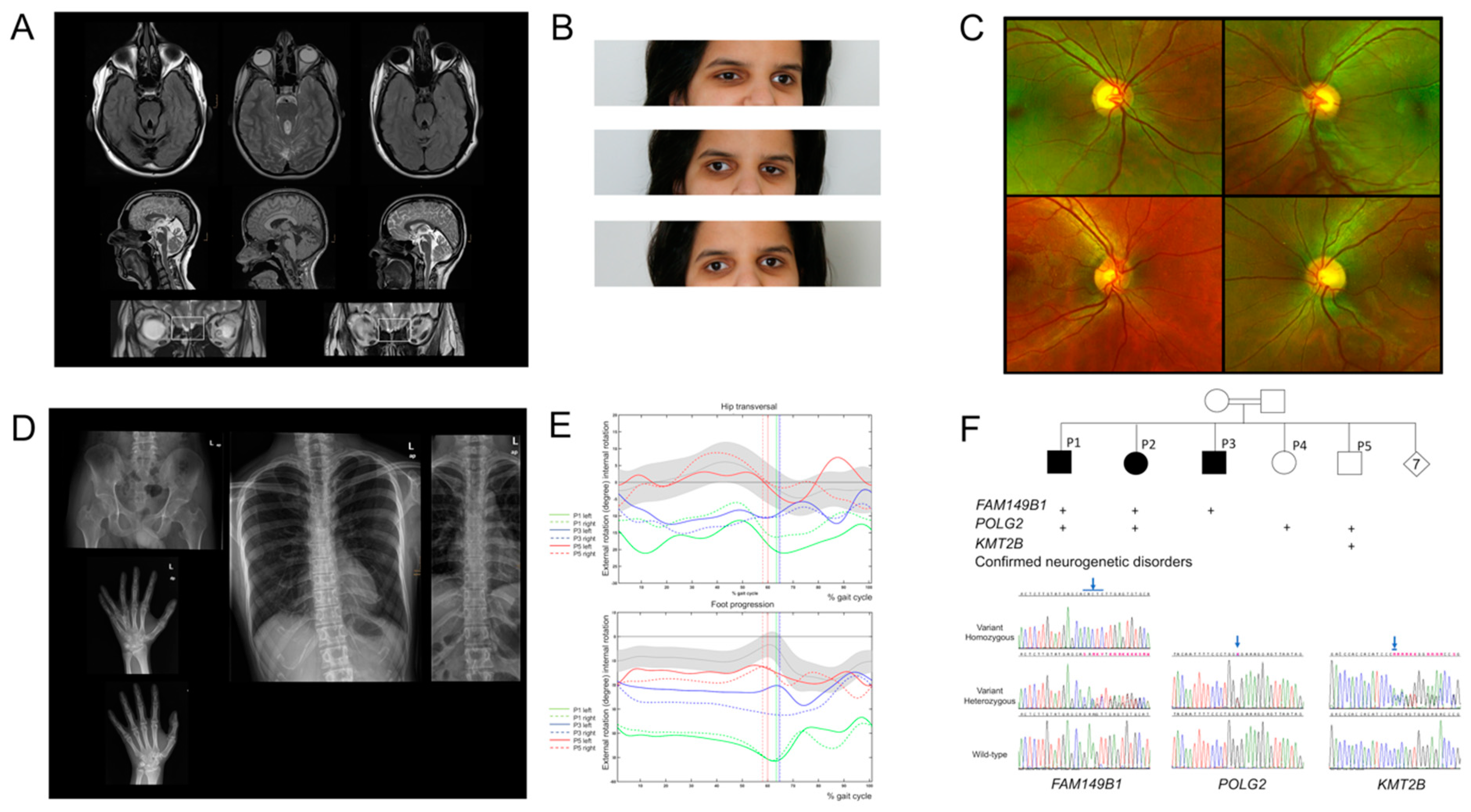
3. Results
3.1. Clinical Characteristics
3.2. Identification of a Homozygous Truncating JS-Causing FAM149B1 Variant
3.3. Identification of Two Additional Neurogenetic Disorders in the Same Family
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisi, M.; Glass, I. Joubert Syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; NCBI: Seattle, WA, USA, 1993. [Google Scholar]
- Poretti, A.; Huisman, T.A.; Scheer, I.; Boltshauser, E. Joubert syndrome and related disorders: Spectrum of neuroimaging findings in 75 patients. AJNR Am. J. Neuroradiol. 2011, 32, 1459–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, D. Joubert syndrome: Insights into brain development, cilium biology, and complex disease. Semin Pediatr Neurol. 2009, 16, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Bachmann-Gagescu, R.; Dempsey, J.C.; Phelps, I.G.; O’Roak, B.J.; Knutzen, D.M.; Rue, T.C.; Ishak, G.E.; Isabella, C.R.; Gorden, N.; Adkins, J.; et al. Joubert syndrome: A model for untangling recessive disorders with extreme genetic heterogeneity. J. Med. Genet. 2015, 52, 514–522. [Google Scholar] [CrossRef] [Green Version]
- Munke, M.; McDonald, D.M.; Cronister, A.; Stewart, J.M.; Gorlin, R.J.; Zackai, E.H. Oral-facial-digital syndrome type VI (Varadi syndrome): Further clinical delineation. Am. J. Med. Genet. 1990, 35, 360–369. [Google Scholar] [CrossRef]
- Varadi, V.; Szabo, L.; Papp, Z. Syndrome of polydactyly, cleft lip/palate or lingual lump, and psychomotor retardation in endogamic gypsies. J. Med. Genet. 1980, 17, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, R.; Jiang, N.; Alzahrani, F.; Ewida, N.; Al-Sheddi, T.; Alobeid, E.; Musaev, D.; Stanley, V.; Hashem, M.; Ibrahim, N.; et al. Bi-allelic Mutations in FAM149B1 Cause Abnormal Primary Cilium and a Range of Ciliopathy Phenotypes in Humans. Am. J. Hum. Genet. 2019, 104, 731–737. [Google Scholar] [CrossRef] [Green Version]
- Baumann, M.; Steichen-Gersdorf, E.; Krabichler, B.; Muller, T.; Janecke, A.R. A recognizable type of syndromic short stature with arthrogryposis caused by bi-allelic SEMA3A loss-of-function variants. Clin. Genet. 2017, 92, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Klee, K.M.C.; Janecke, A.R.; Civan, H.A.; Rosipal, S.; Heinz-Erian, P.; Huber, L.A.; Muller, T.; Vogel, G.F. AP1S1 missense mutations cause a congenital enteropathy via an epithelial barrier defect. Hum. Genet. 2020, 139, 1247–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, B.P.; Zein, W.M.; Thompson, A.H.; Mokhtarzadeh, M.; Doherty, D.A.; Parisi, M.; Glass, I.A.; Malicdan, M.C.; Vilboux, T.; Vemulapalli, M.; et al. Joubert Syndrome: Ophthalmological Findings in Correlation with Genotype and Hepatorenal Disease in 99 Patients Prospectively Evaluated at a Single Center. Ophthalmology 2018, 125, 1937–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulaga, H.M.; Leitch, C.C.; Eichers, E.R.; Badano, J.L.; Lesemann, A.; Hoskins, B.E.; Lupski, J.R.; Beales, P.L.; Reed, R.R.; Katsanis, N. Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat. Genet. 2004, 36, 994–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, J.J.; Noblet, V.; Kremer, S.; Moliere, S.; Dollfus, H.; Marion, V.; Goetz, N.; Muller, J.; Riehm, S. Value of MRI olfactory bulb evaluation in the assessment of olfactory dysfunction in Bardet-Biedl syndrome. Clin. Genet. 2016, 90, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Dahmer-Heath, M.; Schriever, V.; Kollmann, S.; Schleithoff, C.; Titieni, A.; Cetiner, M.; Patzer, L.; Tonshoff, B.; Hansen, M.; Pennekamp, P.; et al. Systematic evaluation of olfaction in patients with hereditary cystic kidney diseases/renal ciliopathies. J. Med. Genet. 2020, 58, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Uytingco, C.R.; Green, W.W.; Martens, J.R. Olfactory Loss and Dysfunction in Ciliopathies: Molecular Mechanisms and Potential Therapies. Curr. Med. Chem. 2019, 26, 3103–3119. [Google Scholar] [CrossRef]
- Weiss, A.H.; Doherty, D.; Parisi, M.; Shaw, D.; Glass, I.; Phillips, J.O. Eye movement abnormalities in Joubert syndrome. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4669–4677. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, M.C. Congenital optic disk anomalies. Surv. Ophthalmol. 1994, 39, 89–112. [Google Scholar] [CrossRef]
- Ware, S.M.; Aygun, M.G.; Hildebrandt, F. Spectrum of clinical diseases caused by disorders of primary cilia. Proc. Am. Thorac. Soc. 2011, 8, 444–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.F.; Kowal, T.J.; Ning, K.; Koo, E.B.; Wu, A.Y.; Mahajan, V.B.; Sun, Y. Review of Ocular Manifestations of Joubert Syndrome. Genes 2018, 9, 605. [Google Scholar] [CrossRef] [Green Version]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. A 2019, 179, 2393–2419. [Google Scholar] [CrossRef]
- Huber, C.; Cormier-Daire, V. Ciliary disorder of the skeleton. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160C, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Schmidts, M. Clinical genetics and pathobiology of ciliary chondrodysplasias. J. Pediatr. Genet. 2014, 3, 46–94. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.C.; Czermin, B.; Muller-Ziermann, S.; Bulst, S.; Stewart, J.D.; Hudson, G.; Schneiderat, P.; Abicht, A.; Holinski-Feder, E.; Lochmuller, H.; et al. Late-onset ptosis and myopathy in a patient with a heterozygous insertion in POLG2. J. Neurol. 2010, 257, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Monies, D.; Abouelhoda, M.; Assoum, M.; Moghrabi, N.; Rafiullah, R.; Almontashiri, N.; Alowain, M.; Alzaidan, H.; Alsayed, M.; Subhani, S.; et al. Lessons Learned from Large-Scale, First-Tier Clinical Exome Sequencing in a Highly Consanguineous Population. Am. J. Hum. Genet. 2019, 104, 1182–1201. [Google Scholar] [CrossRef] [Green Version]
- AlAbdi, L.; Alrashseed, S.; Alsulaiman, A.; Helaby, R.; Imtiaz, F.; Alhamed, M.; Alkuraya, F.S. Residual risk for additional recessive diseases in consanguineous couples. Genet. Med. 2021, 1–7. [Google Scholar] [CrossRef]
- Cif, L.; Demailly, D.; Lin, J.P.; Barwick, K.E.; Sa, M.; Abela, L.; Malhotra, S.; Chong, W.K.; Steel, D.; Sanchis-Juan, A.; et al. KMT2B-related disorders: Expansion of the phenotypic spectrum and long-term efficacy of deep brain stimulation. Brain 2020, 143, 3242–3261. [Google Scholar] [CrossRef] [PubMed]
- Tisch, S.; Kumar, K.R. Pallidal Deep Brain Stimulation for Monogenic Dystonia: The Effect of Gene on Outcome. Front. Neurol. 2020, 11, 630391. [Google Scholar] [CrossRef]

{kind=link}
| Clinical Features | P1 | P2 | P3 | Previously Reported | Total |
|---|---|---|---|---|---|
| Age | 40 y | 25 y | 18 y | 2 y, 3 y, 4 y, 17 y | 2 y to 40 y |
| Sex | m | f | m | m = 4, f = 0 | m = 6, f= 1 |
| Central Nervous System Features | |||||
| MTS | + | + | + | 2/3 | 5/6 |
| Olfactory aplasia | + | + | − | 0/3 | 2/6 |
| Corpus callosal dysgenesis | + | − | + | 0/3 | 2/6 |
| DD/ID | + | + | + | 4/4 | 7/7 |
| OMA/strabismus | + | + | + | 4/4 | 7/7 |
| Ptosis | + | − | − | 4/4 | 5/7 |
| Muscular hypotonia | + | + | + | 1/4 | 4/7 |
| Ataxia | + | + | + | 0/4 | 3/7 |
| Duane Syndrome | + | + | + | 0/4 | 3/7 |
| Nystagmus | + | + | + | 0/4 | 3/7 |
| Progressive ophthalmoplegia | + | + | + | 0/4 | 3/7 |
| Seizures | − | − | − | 1/4 | 1/7 |
| Neonatal breathing abnormalities | − | − | − | 0/4 | 0/7 |
| Skeletal Features | |||||
| Mesoaxial polydactyly | + | − | − | 4/4 | 5/7 |
| Brachy- or clinodactyly V | + | − | + | 1/4 | 3/7 |
| Mild skeletal dysplasia | + | + | + | 0/4 | 3/7 |
| Narrow chest | − | − | − | 1/4 | 1/7 |
| Macrocephaly | − | − | + | 2/4 | 3/7 |
| Disproportionate short stature | − | − | − | 0/4 | 0/7 |
| Orofacial Features | |||||
| Oral clefts | − | − | − | 1/4 | 1/7 |
| Lustered hair, infantile onset | − | − | − | 2/4 | 2/7 |
| Multisystemic Features | |||||
| Cardiac defects | − | − | − | 1/4 | 1/7 |
| Renal involvement | − | − | − | 0/4 | 0/7 |
| Retinal involvement | − | − | − | 0/1 | 0/4 |
| Liver involvement | − | − | − | 0/4 | 0/7 |
| Sensorineuronal deafness | + | − | − | 1/4 | 2/ 7 |
| Lateralization defects | − | + | − | 0/4 | 1/7 |
| Miscellaneous | |||||
| Myopia, cataract | |||||
| P1 | P2 | P3 | P4 | P5 | |
|---|---|---|---|---|---|
| Ciliopathy FAM149B1 | + | + | + | − | − |
| Clues |
| ||||
| Pitfalls |
| ||||
| Mitochondriopathy POLG2 | + | + | − | + | + |
| Clues |
| ||||
| Pitfalls |
| ||||
| Dystonia KMT2B | − | − | − | − | + |
| Clues |
| ||||
| Pitfalls |
| ||||
| Sex | M | F | M | F | M |
| Current age | 40 y | 25 y | 18 y | 26 y | 21 y |
| Ocular features | |||||
| Oculomotor disturbances | |||||
| Strabism congenital onset | + | + | + | − | − |
| Intermittend strabism juvenile onset | − | − | − | + | + |
| Nystagmus, vanishing with age due to PO | + | + | + | − | − |
| Duane syndrome vanishing with age due to PO | + | + | + | − | − |
| PO | + Complete age 35 y | + incomplete age 24 y | + incomplete age 18 y | − | − |
| Ptosis | + 5 mm | − | − | − | − |
| Ocular findings | |||||
| Visual acuity (Snellen decimal) RE/LE | 0.3/0.3 | 0.2/0.5 | 0.6/0.5 | 1.0/1.0 | 0.8/0.8 |
| Refraction (diopters, spherical equivalent) RE/LE | −12.0/−12.0 | +0.5/+0.5 | −0.25/−0.25 | +0.25/+0.25 | −1.0/−1.0 |
| Anterior segment | Mild lens opacification; BE | normal; BE | normal; BE | normal; BE | normal; BE |
| Fundoscopy | Myopic fundus degeneration with thinning of the retinal pigment epithelium and chorioid, peripapillariy myopic conus; BE | Mild situs inversus of the retinal vessels showing nasalward emergence from the optic disc; BE | normal; BE | normal; BE | normal; BE |
| Optical coherence tomography (macula) | Myopic thinning of outer retinal layers and choriocapillaris; BE | normal; BE | normal; BE | normal; BE | normal; BE |
| Full-field ERG (ISCEV Standard) | Dark and light adapted ERGs show subnormal amplitudes with normal timing in keeping with high myopia; BE | normal; BE | normal; BE | normal; BE | not feasible due to dystonia; BE |
| Pattern VEP | Not gradable due to loss of fixation/strabism; BE | normal; BE | normal; BE | normal; BE | not feasible due to dystonia; BE |
| Miscellaneous | Squint surgery; age 22 y | ||||
| Neurological features | |||||
| Movement disorder | |||||
| Cerebellar ataxia | + | + | + | − | − |
| Hypo-/Bradykinesia | + | + | + | − | − |
| Myoclonus | − | − | − | + | − |
| Tremor | − | − | − | − | Dystonic tremor |
| Athetosis/Dystonia | − | + | + | − | +++ |
| Dysarthria | − | + | + | − | + |
| Myopathy or exercise intolerance and exercise induced lactat elevation | + | + | − | + | + |
| Infantile muscular hypotonia | + | + | + | − | − |
| Motor skill delay | + | + | + | − | − |
| Intellectual disability/IQ | +/ND | +/IQ 51 | +/IQ 62 | −/ND | +/IQ 64 |
| Discrepancy between speech comprehension and verbal abilities | + | + | + | − | − |
| GDI score by gait analysisGDI normal range = 100 | 28 | ND | 74 | ND | 94 |
| Skeletal phenotype | |||||
| Mild skeletal dysplasia with abnormal gait | + | + | + | − | − |
| Trident acetabular roofs with protrusion acetabuli | + | + | + | − | − |
| Premature and overt calcification of costal cartilage | + | + | − | − | − |
| Increased hip rotation | + | + | + | − | − |
| Increased external feet rotation | + | + | + | − | − |
| Dysproportionate stature | − | − | − | − | − |
| Macrocephaly | − | − | + | − | − |
| Brain MRI | |||||
| Molar tooth sign | + | + | + | − | − |
| Cerebellar hypoplasia of the upper vermis | + | + | + | − | − |
| Corpus callosal dysgenesis | + | − | + | − | − |
| Olfactory aplasia | + | + | − | − | − |
| Basal ganglia abnormalities | − | − | − | − | + |
| Additional features | |||||
| Endocrine abnormalities | − | − | − | − | − |
| Hearing loss | High frequency spectrum bilateral age 40 y | − | − | + unilateral at age 26 y | + |
| Dysphagia adult onset | ? | − | − | + | + |
| Sleep disturbance | + | − | − | + | − |
| Facial dysmorphism | − | − | − | − | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siegert, S.; Mindler, G.T.; Brücke, C.; Kranzl, A.; Patsch, J.; Ritter, M.; Janecke, A.R.; Vodopiutz, J. Expanding the Phenotype of the FAM149B1-Related Ciliopathy and Identification of Three Neurogenetic Disorders in a Single Family. Genes 2021, 12, 1648. https://doi.org/10.3390/genes12111648
Siegert S, Mindler GT, Brücke C, Kranzl A, Patsch J, Ritter M, Janecke AR, Vodopiutz J. Expanding the Phenotype of the FAM149B1-Related Ciliopathy and Identification of Three Neurogenetic Disorders in a Single Family. Genes. 2021; 12(11):1648. https://doi.org/10.3390/genes12111648
Chicago/Turabian StyleSiegert, Sandy, Gabriel T. Mindler, Christof Brücke, Andreas Kranzl, Janina Patsch, Markus Ritter, Andreas R. Janecke, and Julia Vodopiutz. 2021. "Expanding the Phenotype of the FAM149B1-Related Ciliopathy and Identification of Three Neurogenetic Disorders in a Single Family" Genes 12, no. 11: 1648. https://doi.org/10.3390/genes12111648
APA StyleSiegert, S., Mindler, G. T., Brücke, C., Kranzl, A., Patsch, J., Ritter, M., Janecke, A. R., & Vodopiutz, J. (2021). Expanding the Phenotype of the FAM149B1-Related Ciliopathy and Identification of Three Neurogenetic Disorders in a Single Family. Genes, 12(11), 1648. https://doi.org/10.3390/genes12111648