
Mitochondrial Strokes: Diagnostic Challenges and Chameleons

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
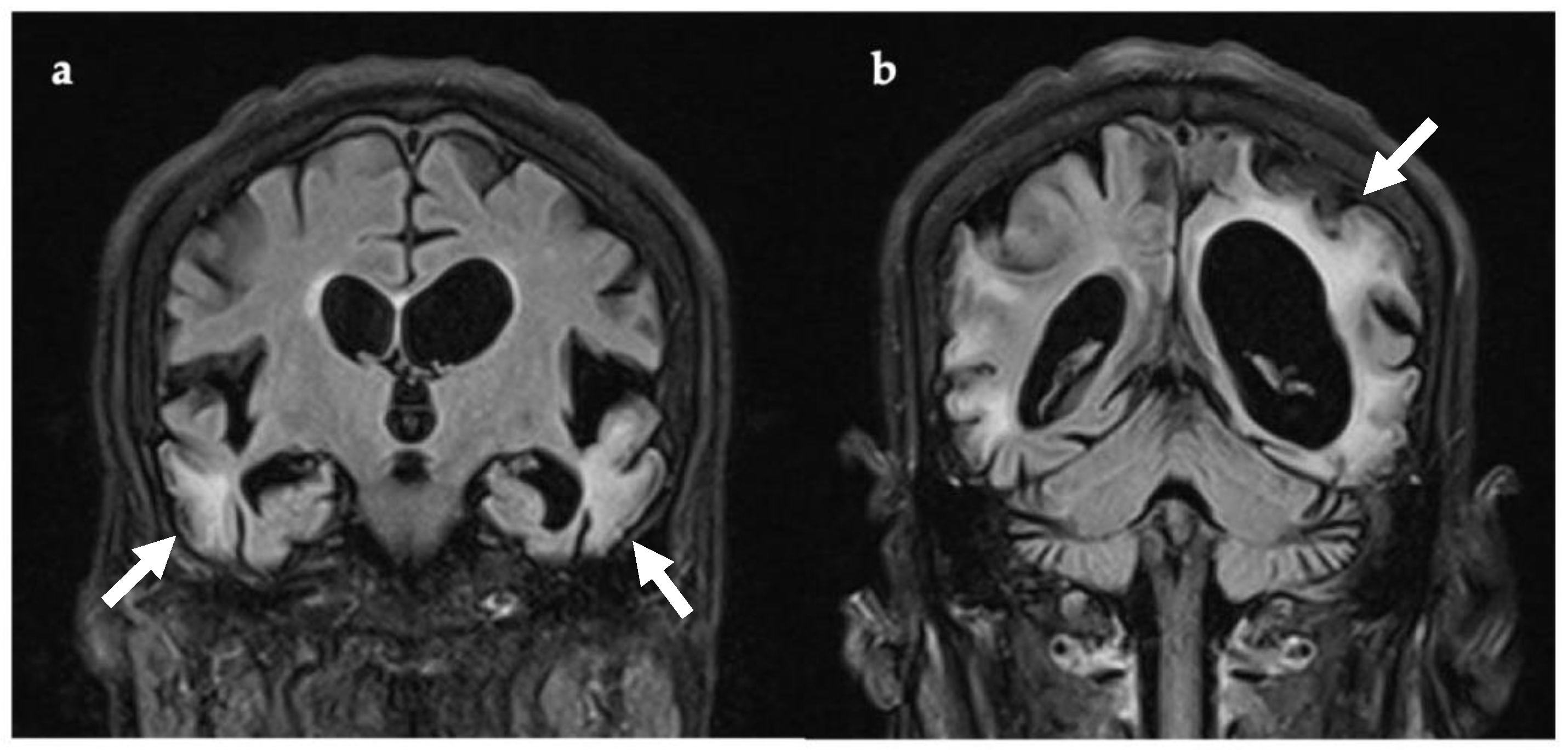
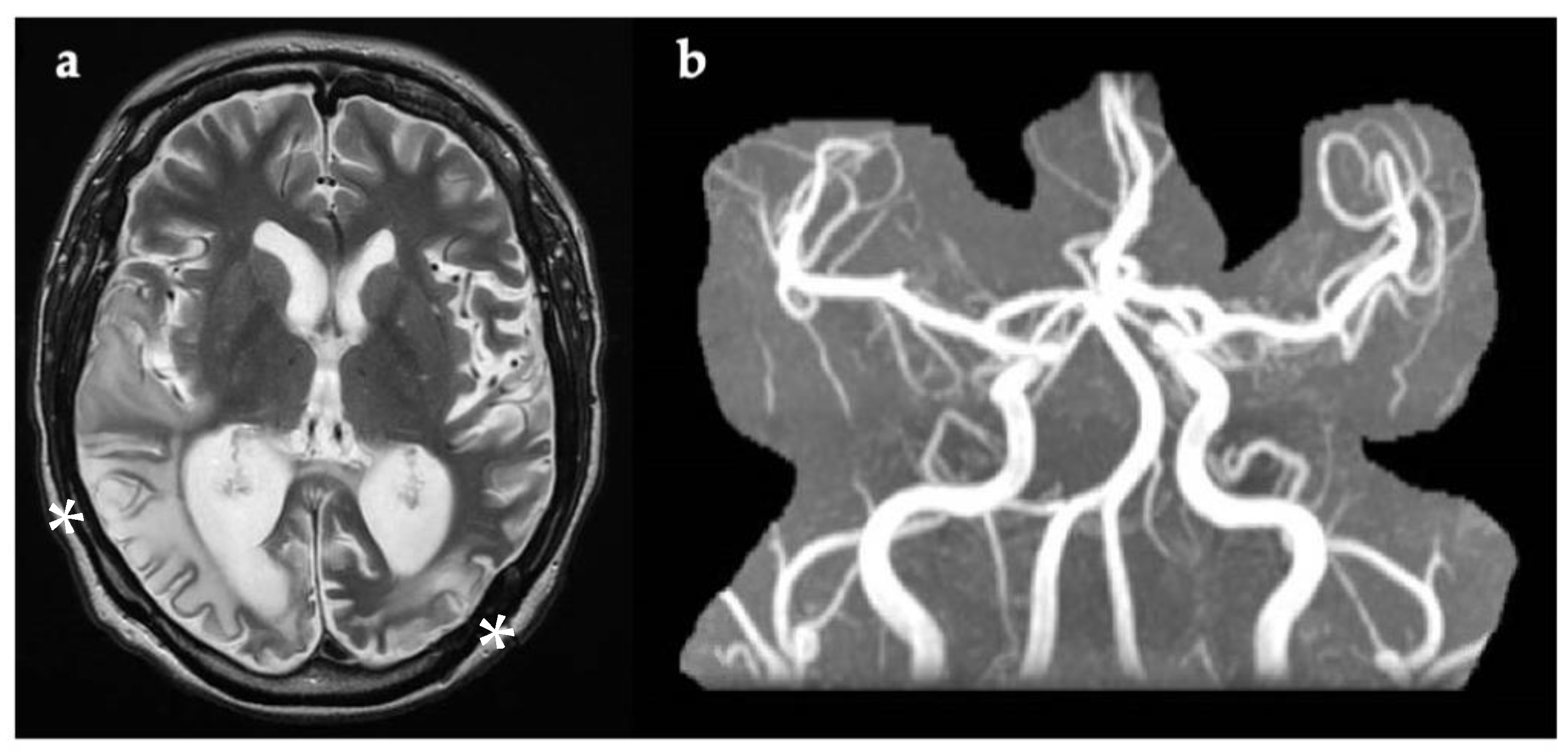
3.1. Case 1
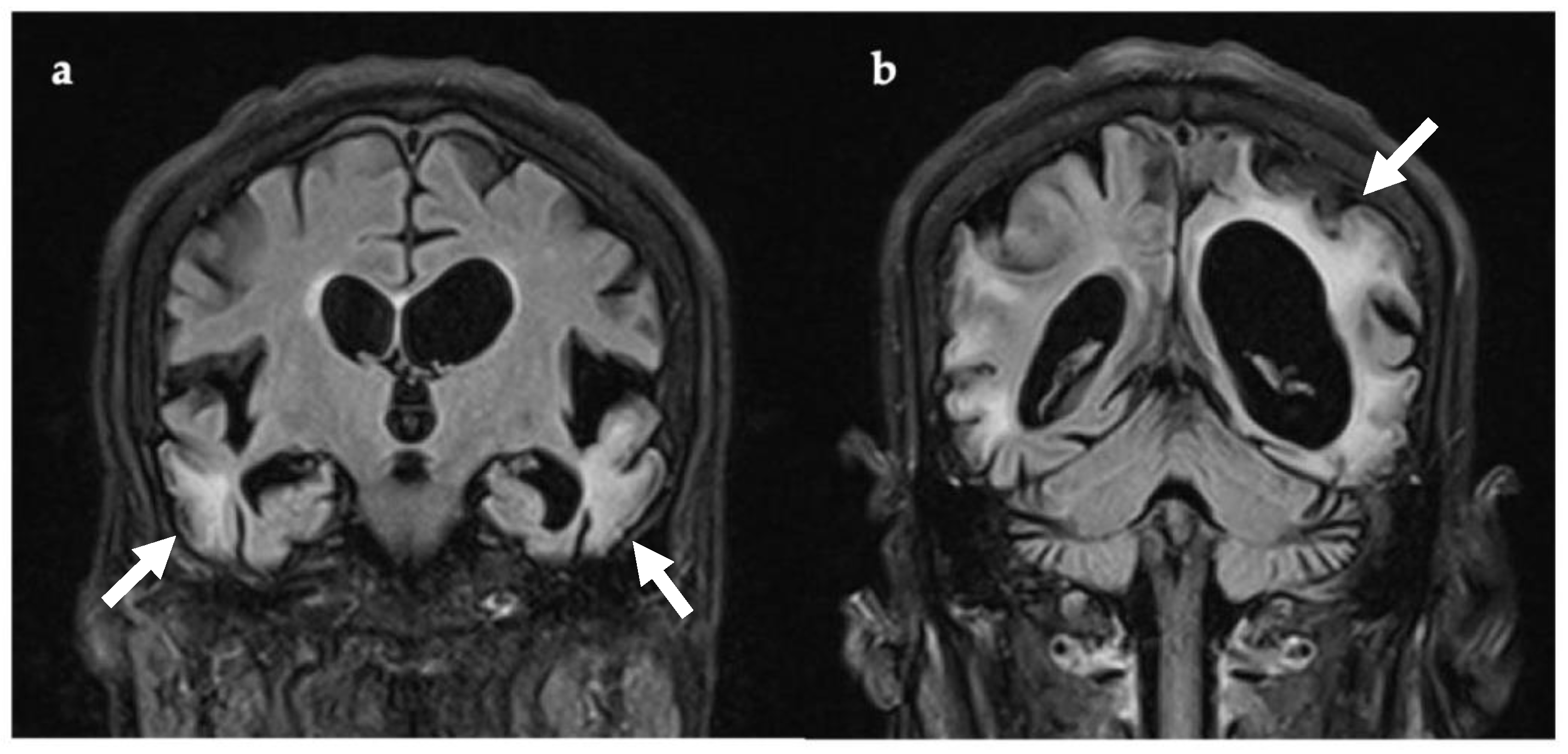
3.2. Case 2
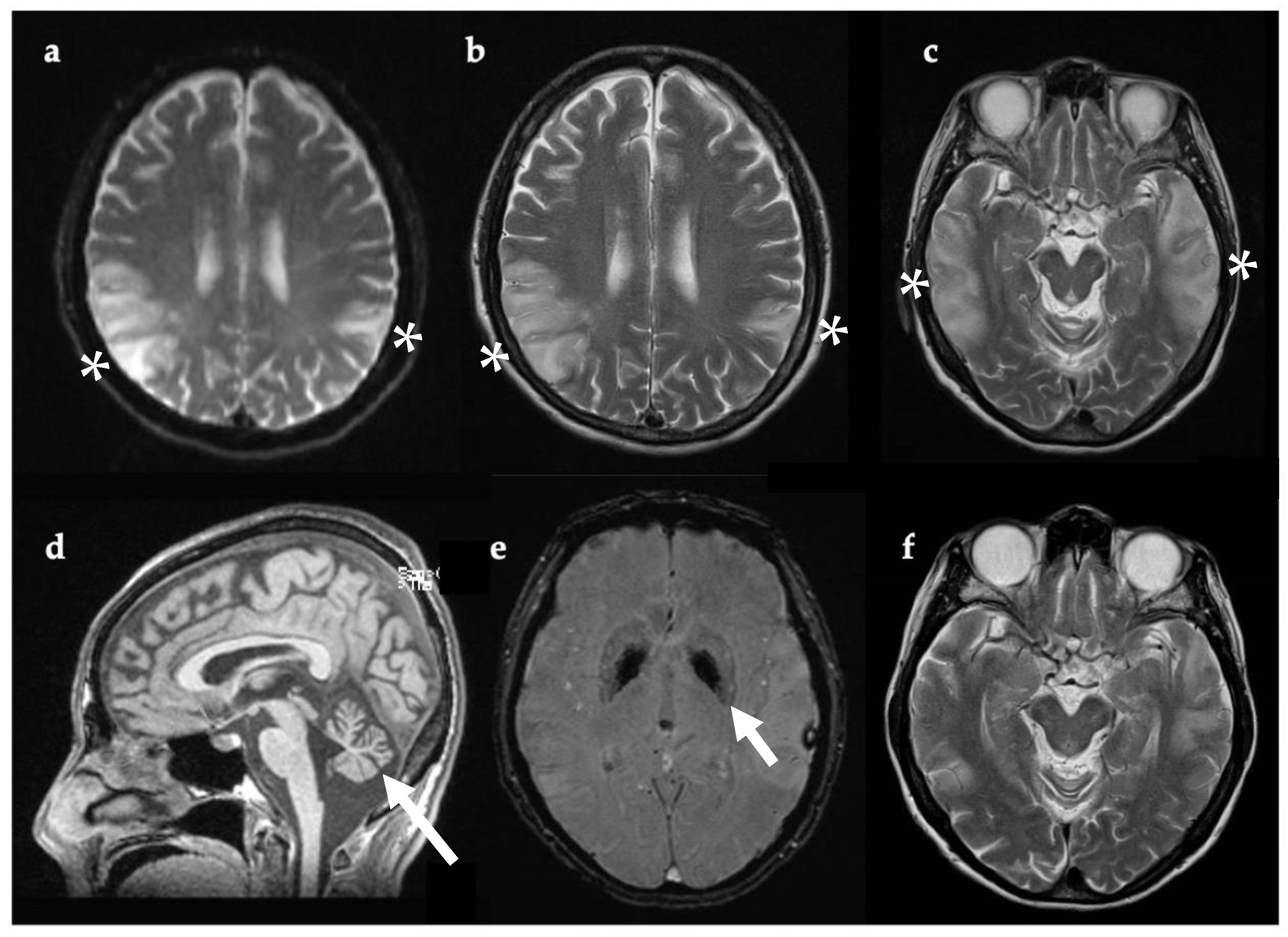
3.3. Case 3
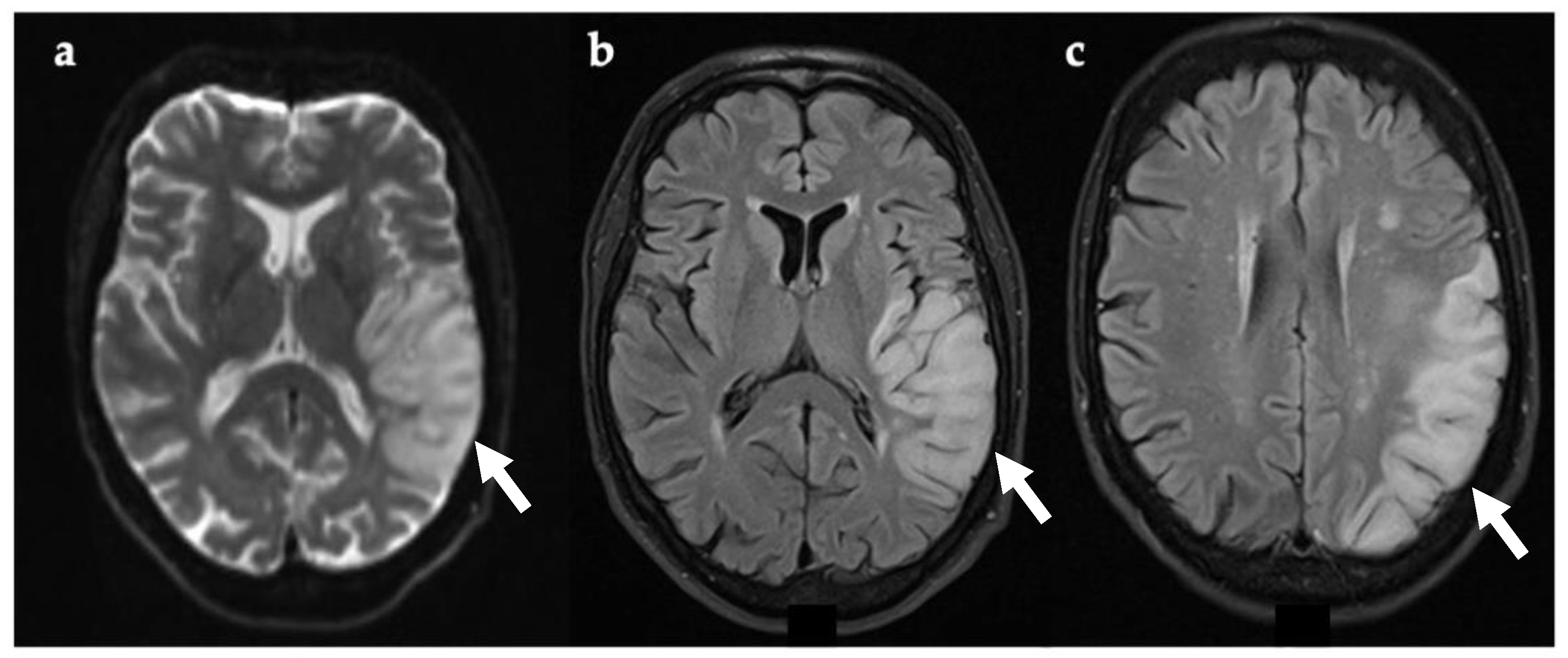
3.4. Case 4
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ng, Y.S.; Bindoff, L.A.; Gorman, G.S.; Horvath, R.; Klopstock, T.; Mancuso, M.; Martikainen, M.H.; McFarland, R.; Nesbitt, V.; Pitceathly, R.D.S.; et al. Consensus-based statements for the management of mitochondrial stroke-like episodes. Wellcome Open Res. 2019, 4, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, Y.; Nonaka, I.; Horai, S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Shanske, S.; Coku, J.; Lu, J.; Ganesh, J.; Krishna, S.; Tanji, K.; Bonilla, E.; Naini, A.B.; Hirano, M.; DiMauro, S. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome: Evidence from 12 cases. Arch. Neurol. 2008, 65, 368–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisca, G.; Fiorillo, C.; Nesti, C.; Trucco, F.; Derchi, M.; Andaloro, A.; Assereto, S.; Morcaldi, G.; Pedemonte, M.; Minetti, C.; et al. Early onset cardiomyopathy associated with the mitochondrial tRNALeu((UUR)) 3271T > C MELAS mutation. Biochem. Biophys. Res. Commun. 2015, 458, 601–604. [Google Scholar] [CrossRef]
- Deschauer, M.; Tennant, S.; Rokicka, A.; He, L.; Kraya, T.; Turnbull, D.M.; Zierz, S.; Taylor, R.W. MELAS associated with mutations in the POLG1 gene. Neurology 2007, 68, 1741–1742. [Google Scholar] [CrossRef]
- Kristen, M.; Laricchia, N.J.L.; Nicholas, A.; Watts, N.A.; Shand, M.; Haessly, A.; Gauthier, L.; Benjamin, D.; Banks, E.; Soto, J.; et al. Mitochondrial DNA variation across 56,434 individuals in gnomAD. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yatsuga, S.; Povalko, N.; Nishioka, J.; Katayama, K.; Kakimoto, N.; Matsuishi, T.; Koga, Y.; Taro Matsuoka for MELAS Study Group in Japan. MELAS: A nationwide prospective cohort study of 96 patients in Japan. Biochim. Biophys. Acta 2012, 1820, 619–624. [Google Scholar] [CrossRef]
- Tschampa, H.J.; Urbach, H.; Greschus, S.; Kunz, W.S.; Kornblum, C. Neuroimaging characteristics in mitochondrial encephalopathies associated with the m.3243A > G MTTL1 mutation. J. Neurol. 2013, 260, 1071–1080. [Google Scholar] [CrossRef]
- Lax, N.Z.; Gorman, G.S.; Turnbull, D.M. Review: Central nervous system involvement in mitochondrial disease. Neuropathol. Appl. Neurobiol. 2017, 43, 102–118. [Google Scholar] [CrossRef]
- Valencia-Sanchez, C.; Pittock, S.J.; Mead-Harvey, C.; Dubey, D.; Flanagan, E.P.; Lopez-Chiriboga, S.; Trenerry, M.R.; Zalewski, N.L.; Zekeridou, A.; McKeon, A. Brain dysfunction and thyroid antibodies: Autoimmune diagnosis and misdiagnosis. Brain Commun. 2021, 3, fcaa233. [Google Scholar] [CrossRef]
- Zhang, W.; Yan, L.; Jiao, J. Repeated misdiagnosis of a relapsed atypical anti-NMDA receptor encephalitis without an associated ovarian teratoma. Neurosci. Lett. 2017, 638, 135–138. [Google Scholar] [CrossRef]
- Sharfstein, S.R.; Gordon, M.F.; Libman, R.B.; Malkin, E.S. Adult-onset MELAS presenting as herpes encephalitis. Arch. Neurol. 1999, 56, 241–243. [Google Scholar] [CrossRef]
- McClelland, G.; Rodgers, H.; Flynn, D.; Price, C.I. The frequency, characteristics and aetiology of stroke mimic presentations: A narrative review. Eur. J. Emerg. Med. 2019, 26, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Poole, O.V.; Everett, C.M.; Gandhi, S.; Marino, S.; Bugiardini, E.; Woodward, C.; Lam, A.; Quinlivan, R.; Hanna, M.G.; Pitceathly, R.D.S. Adult-onset Leigh syndrome linked to the novel stop codon mutation m.6579G > A in MT-CO1. Mitochondrion 2019, 47, 294–297. [Google Scholar] [CrossRef] [Green Version]
- Hirano, M.; Ricci, E.; Koenigsberger, M.R.; Defendini, R.; Pavlakis, S.G.; DeVivo, D.C.; DiMauro, S.; Rowland, L.P. Melas: An original case and clinical criteria for diagnosis. Neuromuscul. Disord. 1992, 2, 125–135. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, V.; Pitceathly, R.D.; Turnbull, D.M.; Taylor, R.W.; Sweeney, M.G.; Mudanohwo, E.E.; Rahman, S.; Hanna, M.G.; McFarland, R. The UK MRC Mitochondrial Disease Patient Cohort Study: Clinical phenotypes associated with the m.3243A > G mutation--implications for diagnosis and management. J. Neurol. Neurosurg. Psychiatry 2013, 84, 936–938. [Google Scholar] [CrossRef] [PubMed]
- De Laat, P.; Rodenburg, R.R.; Roeleveld, N.; Koene, S.; Smeitink, J.A.; Janssen, M.C. Six-year prospective follow-up study in 151 carriers of the mitochondrial DNA 3243 A > G variant. J. Med. Genet. 2021, 58, 48–55. [Google Scholar] [CrossRef] [PubMed]
- De Laat, P.; Koene, S.; van den Heuvel, L.P.; Rodenburg, R.J.; Janssen, M.C.; Smeitink, J.A. Clinical features and heteroplasmy in blood, urine and saliva in 34 Dutch families carrying the m.3243A > G mutation. J. Inherit. Metab. Dis. 2012, 35, 1059–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marotta, R.; Reardon, K.; McKelvie, P.A.; Chiotis, M.; Chin, J.; Cook, M.; Collins, S.J. Association of the MELAS m.3243A > G mutation with myositis and the superiority of urine over muscle, blood and hair for mutation detection. J. Clin. Neurosci. 2009, 16, 1223–1225. [Google Scholar] [CrossRef]
- Frederiksen, A.L.; Andersen, P.H.; Kyvik, K.O.; Jeppesen, T.D.; Vissing, J.; Schwartz, M. Tissue specific distribution of the 3243A- > G mtDNA mutation. J. Med. Genet. 2006, 43, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Finsterer, J. Manifestations of the mitochondrial A3243G mutation. Int. J. Cardiol. 2009, 137, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Paramasivam, A.; Venkatapathi, C.; Sandeep, G.; Meena, A.K.; Uppin, M.S.; Mohapatra, S.; Pitceathly, R.D.S.; Thangaraj, K. Homozygous R627W mutations in POLG cause mitochondrial DNA depletion leading to encephalopathy, seizures and stroke-like episodes. Mitochondrion 2019, 48, 78–83. [Google Scholar] [CrossRef]
- Rajakulendran, S.; Pitceathly, R.D.; Taanman, J.W.; Costello, H.; Sweeney, M.G.; Woodward, C.E.; Jaunmuktane, Z.; Holton, J.L.; Jacques, T.S.; Harding, B.N.; et al. A Clinical, Neuropathological and Genetic Study of Homozygous A467T POLG-Related Mitochondrial Disease. PLoS ONE 2016, 11, e0145500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, D.H.; Choi, Y.C.; Nam, D.E.; Choi, S.S.; Kim, J.W.; Choi, B.O.; Chung, K.W. Identification of FASTKD2 compound heterozygous mutations as the underlying cause of autosomal recessive MELAS-like syndrome. Mitochondrion 2017, 35, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; D’Souza, A.R.; Dallabona, C.; Lodi, T.; Rebelo-Guiomar, P.; Rorbach, J.; Donati, M.A.; Procopio, E.; Montomoli, M.; Guerrini, R.; et al. Defective mitochondrial rRNA methyltransferase MRM2 causes MELAS-like clinical syndrome. Hum. Mol. Genet. 2017, 26, 4257–4266. [Google Scholar] [CrossRef]
- Smith, K.; Chiu, S.; Hunt, C.; Chandregowda, A.; Babovic-Vuksanovic, D.; Keegan, B.M. Late-onset Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like Episodes Presenting With Auditory Agnosia. Neurologist 2019, 24, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, D.; Mauri, E.; Magri, F.; Velardo, D.; Meneri, M.; Abati, E.; Brusa, R.; Faravelli, I.; Piga, D.; Ronchi, D.; et al. Can Intestinal Pseudo-Obstruction Drive Recurrent Stroke-Like Episodes in Late-Onset MELAS Syndrome? A Case Report and Review of the Literature. Front. Neurol. 2019, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Ueki, K.; Wakisaka, Y.; Nakamura, K.; Shono, Y.; Wada, S.; Yoshikawa, Y.; Matsukuma, Y.; Uchiumi, T.; Kang, D.; Kitazono, T.; et al. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes due to m.3243A > G mutation in a 76-year-old woman. J. Neurol. Sci. 2020, 412, 116791. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Wu, X.; Zhu, B.; Yu, J.; Yang, B.; Shi, J. The pooled incidence of post-stroke seizure in 102 008 patients. Top. Stroke Rehabil. 2015, 22, 460–467. [Google Scholar] [CrossRef]
- Li, J.; Zhang, W.; Cui, Z.; Li, Z.; Jiang, T.; Meng, H. Epilepsy Associated With Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes. Front. Neurol. 2021, 12, 675816. [Google Scholar] [CrossRef]
- Lee, H.N.; Eom, S.; Kim, S.H.; Kang, H.C.; Lee, J.S.; Kim, H.D.; Lee, Y.M. Epilepsy Characteristics and Clinical Outcome in Patients With Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes (MELAS). Pediatr. Neurol. 2016, 64, 59–65. [Google Scholar] [CrossRef]
- Galnares-Olalde, J.A.; Lopez-Hernandez, J.C.; Benitez-Alonso, E.O.; de Montellano, D.J.D.; May-Mas, R.N.; Briseno-Godinez, M.E.; Perez-Valdez, E.Y.; Perez-Jovel, E.; Fernandez-Valverde, F.; Leon-Manriquez, E.; et al. Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Episodes (MELAS) Syndrome: Frequency, Clinical Features, Imaging, Histopathologic, and Molecular Genetic Findings in a Third-level Health Care Center in Mexico. Neurologist 2021, 26, 143–148. [Google Scholar] [CrossRef]
- Calabrese, V.P.; Gruemer, H.D.; James, K.; Hranowsky, N.; DeLorenzo, R.J. Cerebrospinal fluid lactate levels and prognosis in status epilepticus. Epilepsia 1991, 32, 816–821. [Google Scholar] [CrossRef]
- Terent, A.; Ronquist, G. Cerebrospinal fluid markers of disturbed brain cell metabolism in patients with stroke and global cerebral ischemia. Acta Neurol. Scand. 1980, 62, 327–335. [Google Scholar] [CrossRef]
- Kostulas, N.; Larsson, M.; Kall, T.B.; von Euler, M.; Nathanson, D. Safety of thrombolysis in stroke mimics: An observational cohort study from an urban teaching hospital in Sweden. BMJ Open 2017, 7, e016311. [Google Scholar] [CrossRef]
- Tetsuka, S.; Ogawa, T.; Hashimoto, R.; Kato, H. Clinical features, pathogenesis, and management of stroke-like episodes due to MELAS. Metab. Brain Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, C.; Galanaud, D.; Samson, Y.; Sahel, M.; Dormont, D.; Wechsler, B.; Marsault, C. Can diffusion weighted magnetic resonance imaging help differentiate stroke from stroke-like events in MELAS? J. Neurol. Neurosurg. Psychiatry 2000, 69, 248–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jose da Rocha, A.; Tulio Braga, F.; Carlos Martins Maia, A., Jr.; Jorge da Silva, C.; Toyama, C.; Pereira Pinto Gama, H.; Kok, F.; Rodrigues Gomes, H. Lactate detection by MRS in mitochondrial encephalopathy: Optimization of technical parameters. J. Neuroimaging 2008, 18, 1–8. [Google Scholar] [CrossRef]
- Iizuka, T.; Sakai, F. Pathogenesis of stroke-like episodes in MELAS: Analysis of neurovascular cellular mechanisms. Curr. Neurovasc. Res. 2005, 2, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Salsaa, M.; Pereira, B.; Liu, J.; Yu, W.; Jadhav, S.; Huttemann, M.; Greenberg, M.L. Valproate inhibits mitochondrial bioenergetics and increases glycolysis in Saccharomyces cerevisiae. Sci. Rep. 2020, 10, 11785. [Google Scholar] [CrossRef] [PubMed]
- Johns, D.R.; Stein, A.G.; Wityk, R. MELAS syndrome masquerading as herpes simplex encephalitis. Neurology 1993, 43, 2471–2473. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.C.; Yang, F.C.; Perng, C.L.; Tso, A.C.; Wong, L.J.; Hsu, C.H. Adult-onset of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome presenting as acute meningoencephalitis: A case report. J. Emerg. Med. 2012, 43, e163–e166. [Google Scholar] [CrossRef] [PubMed]
- Gooriah, R.; Dafalla, B.E.; Venugopalan, T.C. Led astray: MELAS initially misdiagnosed as herpes simplex encephalitis. Acta Neurol. Belg. 2015, 115, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Tang, M.X.; Gorman, G.S.; De Sutter, P.; Heindryckx, B. Novel reproductive technologies to prevent mitochondrial disease. Hum. Reprod. Update 2017, 23, 501–519. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Het 1 |
Age of 1st SLE (Latency to PMD Diagnosis) | Neurological Presentation of SLE | Brain MRI | EEG |
Other PMD Symptoms |
Differential Diagnosis | ||
| Days 2 | Findings | Days 2 | Findings | ||||||
| 1 | B: neg U: 29% | 51 y (10 y) | Imbalance, drowsiness, seizures | 21 | Lesions in temporo-parieto-occipital lobes bilaterally | 15 | Occasional left temporal sharp waves | Epilepsy, diabetes, deafness | Viral encephalitis |
| 2 | B: 6% M: 74% | 51 y (6 m) | Headache, confusion, visual hallucination, behavioural changes | 11 | Bilateral temporo-parietal cortical lesions, pallidal mineralisation and cerebellar atrophy | 7 | Periodic right temporal waves | Diabetes | Autoimmune/ Viral encephalitis |
| 3 | B: 23% | 37 y (PMD diagnosed before) | Expressive aphasia, seizures | 1 | Left temporal lobe lesion | 10 | Slow activity on the left hemisphere | Hypertrophic CM, diabetes | Left MCA infarct |
| 4 | B: 5% U: 46% Br: 63% | 35 y (9 y) | Left-sided UMN VIIth cranial nerve palsy. LUL weakness, seizures | 4 | Multiple areas of white matter and cortical increased T2w signal within the posterior cerebral hemispheres | 3 | Spike and waves and sharp waves over the right frontal areas | None | CNS vasculitis |
| Clinical Features | Radiological Features |
|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pizzamiglio, C.; Bugiardini, E.; Macken, W.L.; Woodward, C.E.; Hanna, M.G.; Pitceathly, R.D.S. Mitochondrial Strokes: Diagnostic Challenges and Chameleons. Genes 2021, 12, 1643. https://doi.org/10.3390/genes12101643
Pizzamiglio C, Bugiardini E, Macken WL, Woodward CE, Hanna MG, Pitceathly RDS. Mitochondrial Strokes: Diagnostic Challenges and Chameleons. Genes. 2021; 12(10):1643. https://doi.org/10.3390/genes12101643
Chicago/Turabian StylePizzamiglio, Chiara, Enrico Bugiardini, William L. Macken, Cathy E. Woodward, Michael G. Hanna, and Robert D. S. Pitceathly. 2021. "Mitochondrial Strokes: Diagnostic Challenges and Chameleons" Genes 12, no. 10: 1643. https://doi.org/10.3390/genes12101643
APA StylePizzamiglio, C., Bugiardini, E., Macken, W. L., Woodward, C. E., Hanna, M. G., & Pitceathly, R. D. S. (2021). Mitochondrial Strokes: Diagnostic Challenges and Chameleons. Genes, 12(10), 1643. https://doi.org/10.3390/genes12101643