
High-Dimensional Single-Cell Transcriptomics in Melanoma and Cancer Immunotherapy

 ,
,  and
and
Abstract
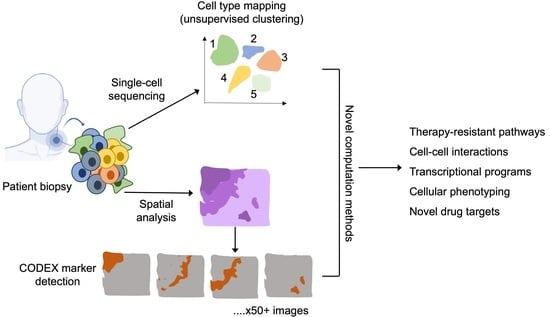
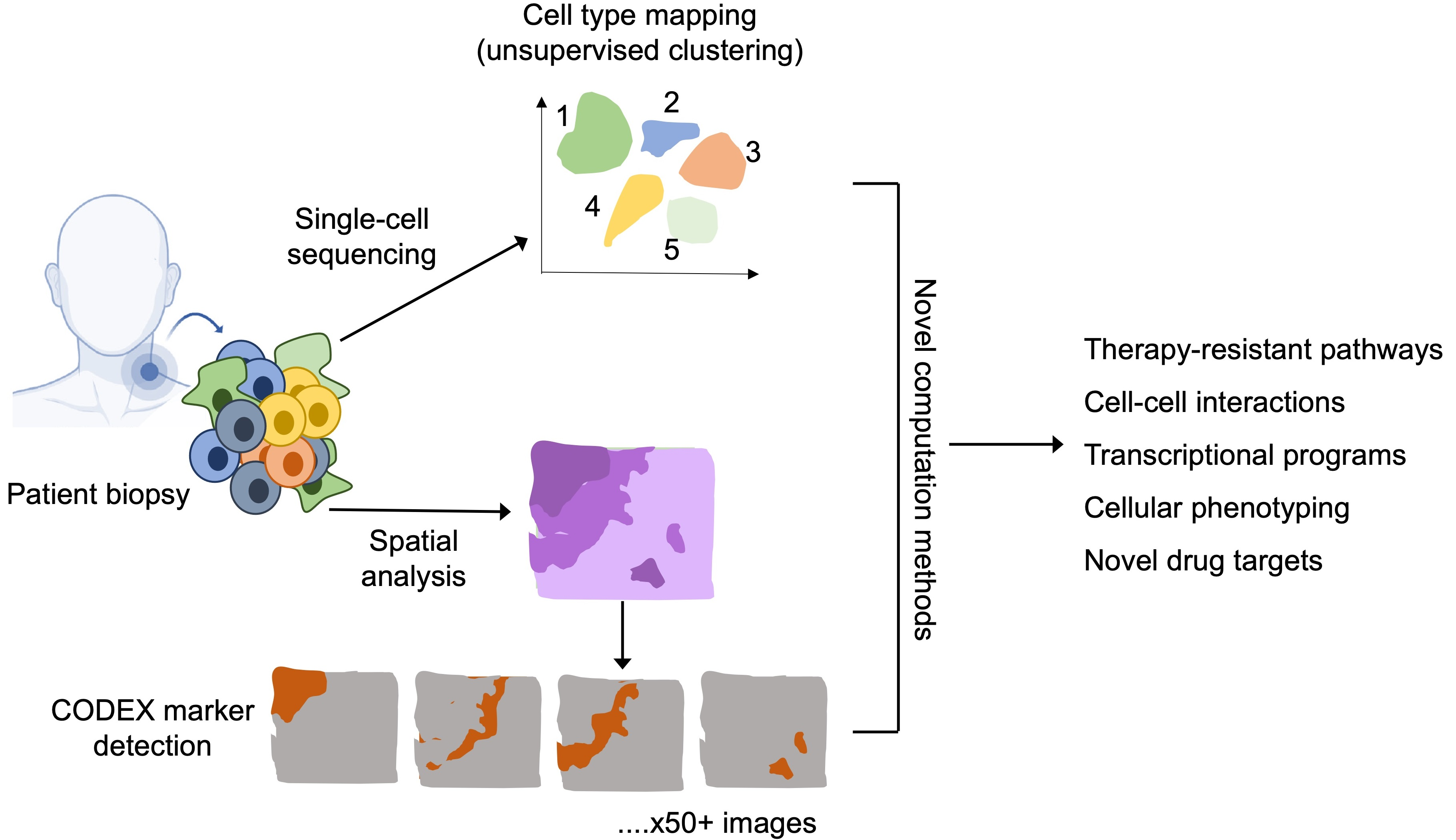
:
1. Introduction
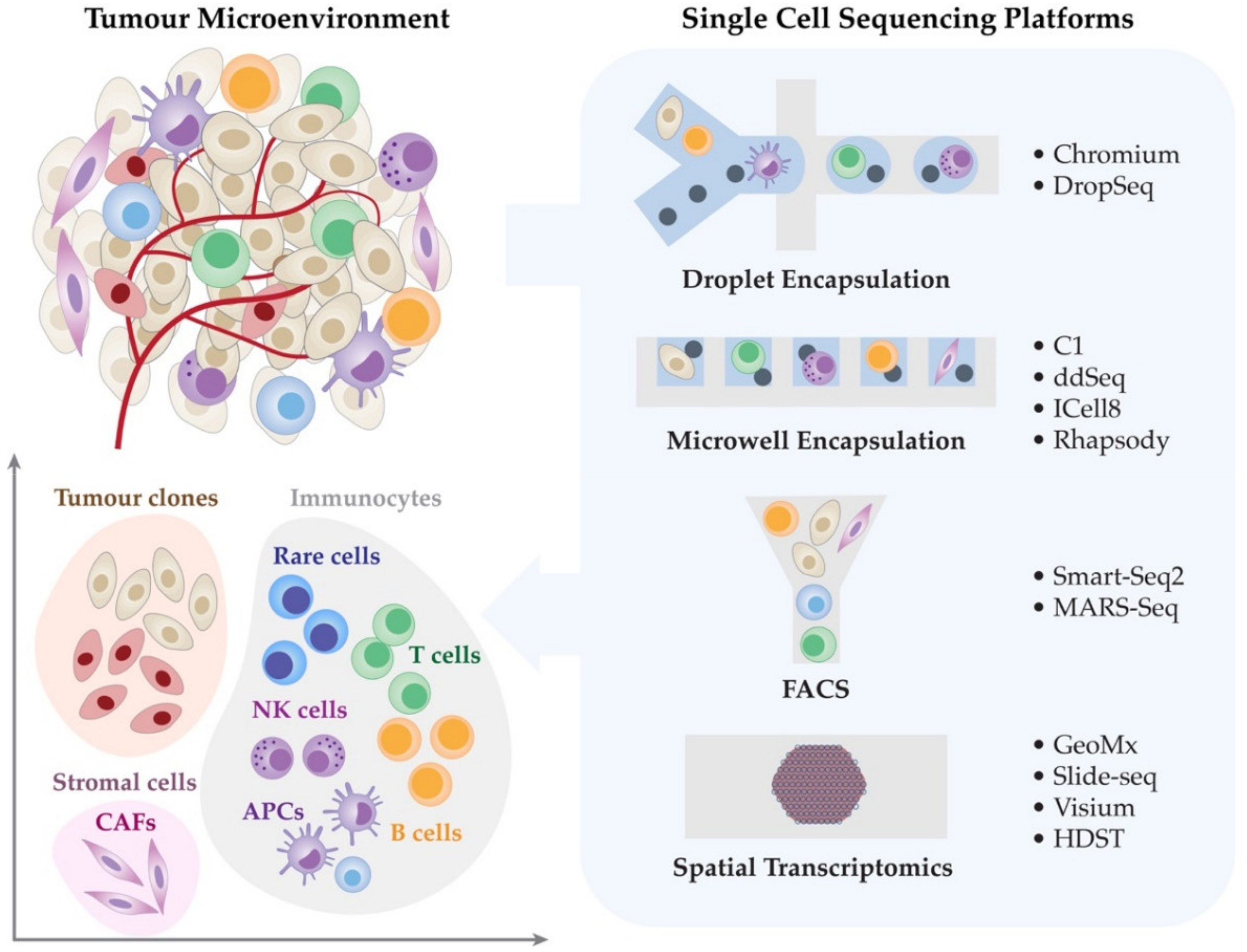
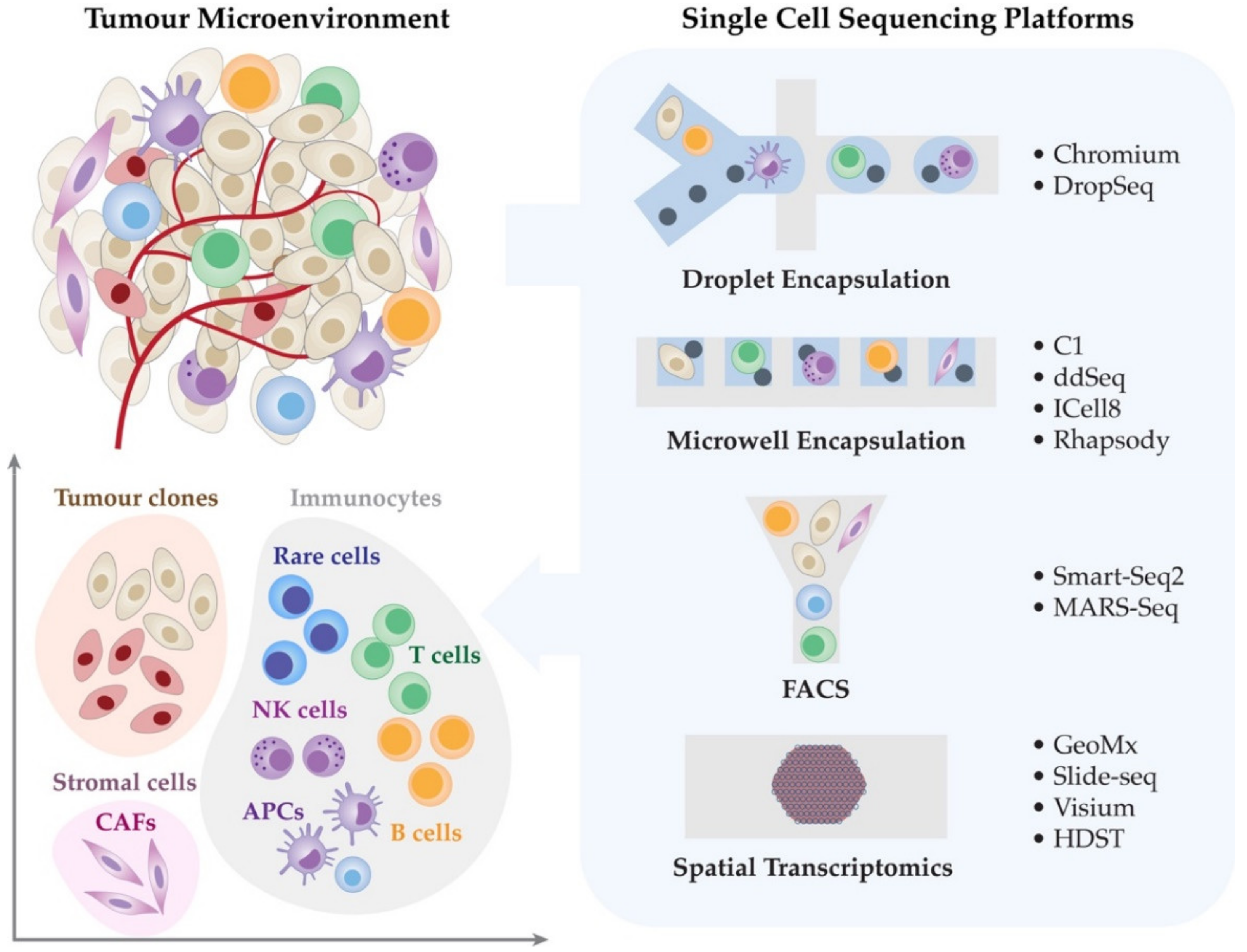
2. Single-Cell Transcriptomic Technologies
2.1. Droplet Encapsulation Technologies
2.2. Microwell Encapsulation Platforms
2.3. Fluorescence-Activated Cell Sorting (FACS)

{kind=link}
{kind=link}
| Platform | Company/Academic | Method of Single-Cell Capture | Capture Efficiency | Doublet Rate | Number of Captured Cells | Cell Size Restrictions | Analytical Tool | Advantages | Relative Limitations | References |
|---|---|---|---|---|---|---|---|---|---|---|
| Chromium | 10x Genomics | Droplet encapsulation | 65% | 0.90% | 100–80,000 | Independent of cell size, but generally up to 50 µm | 10x analysis suite including Cell Ranger and Loupe Browser; Seurat R package | Easy to operate; cost effective; intensive support for end-to-end solution; flexible options for multiple applications | High concentration of viable cells required; Little control over cell input | [59,78] |
| DropSeq (Nadia) | Dolomite-bio | Droplet encapsulation | 10% | 1.80–11.3% | 103–104 | None for mammalian cells | Open platform | High throughput; low cost | High concentration of viable cells required; low cell capture efficiency; skills required to operate; minimal support for data processing and analysis. | [60,61] |
| C1 | Fluidigm | Microwell encapsulation | 39% | 3–30% | 96 or 800 | 5–10, 10–17, or 17–25 µm | Fluidigm Singular Analysis Toolset Software | Full-length transcript; customisable workflow (able to exclude empty wells and doublets) | Limited cell capture; low throughput (up to 96 or 800 cells); high cost of cartridges; relatively long preparation time (two runs per day); fresh tissue or cells required | [56,82] |
| ddSeq | Illumina/Bio-Rad | Microwell encapsulation | 3–4% | 5.80% | 103–104 | None for mammalian cells | Illumina BaseSpace or ddSeeker R package | Easy to operate; flexibility of kits for different number of cells; intensive support for end-to-end solution | High concentration of viable cells required; no users modification; single application (RNA-seq) | [56,78] |
| ICell8 | Takara-Bio | Microwell encapsulation | 37% | 1.3–4% | 1800 | 5–100 μm | CELLSTUDIO software | Easy to operate; full-length transcript; customisable workflow (able to exclude empty wells and doublets) | Specialised bioinformatic tools required; single application (RNA-seq) | [62,78] |
| Rhapsody | BD Biosciences | Microwell encapsulation | 65% | 2–10% | 100–40,000 | 5 to 30 μm | BD Rhapsody Analysis Pipelines and SeqGeq Software | Easy to operate; intensive support for end-to-end solution; simultaneously measure protein and mRNA expression; optimise costs based on subsampling and targeted panels | Low sequencing throughput; custom panel of up to 500 targets | [56,66,67,83] |
| Smart-Seq2 | [75,76] | FACS | 80% | 1% | No limitation | None for mammalian cells | Open platform | No limitations of cell size, shape or homogeneity; simultaneously measure DNA and RNA; high practicality (uses off the shelf reagents); full-length transcript | No options for barcoding and UMI (no multiplexing and gene quantification of samples); laborious worflow due to numerous pipetting steps | [74,75,84] |
| MARS-Seq | [77] | FACS | 92% | 2% | No limitation | None for mammalian cells | Open platform | Automated process; suitable for rare cell sorting; No limitations of cell size, shape or homogeneity | Specialised bioinformatic tools required | [76,78] |
3. Spatially Resolved RNA Technologies
3.1. NanoString GeoMx Digital Spatial Profiler (DSP)
3.2. 10x Genomics Visium
3.3. Slide-Seq
3.4. High-Definition Spatial Transcriptomics (HDST)
| Platform | Company/Academic | Detection Efficiency | Resolution | Number of Captured Cells | Sample Type | Analytical Tool | Advantages | Relative Limitations | References |
|---|---|---|---|---|---|---|---|---|---|
| GeoMx | NanoString | Not reported | 10–600 μm | 20–200 cells per ROI | Fresh-frozen or FFPE | GeoMx Data Centre Software | Easy to operate (high level of automation); intensive support for end-to-end solution; Ability to profile protein/RNA; single-cell level | Low efficiency of cell capture when using smaller ROIs; Require user-defined ROIs | [91] |
| Slide-seq | [87] | 0.30% | 10 μm | ~70,000 | Fresh-frozen | Open platform or Seurat R package | Relatively high resolution; scalability; spatial resolution for large tissue volumes | Low sensitivity; minimal support for data processing and analysis | [90] |
| Visium | 10x Genomics | >6.9% | 55 μm | 1–10 cells per ROI | Fresh-frozen or FFPE | 10x Space Ranger | Intensive support for end-to-end solution; coverage across a large area of tissue | User-defined regions contain multiple cells | [99] |
| High-definition spatial transcriptomics | [89] | 1.30% | 2 μm | ~160,000 | Fresh-frozen | Open platform | High resolution | Low sensitivity; minimal support for data processing and analysis | [92] |
4. Dissecting the Tumour Immune Microenvironment Using Single-Cell Approaches
4.1. Dissecting Intra-Tumoural Heterogeneity (ITH)
4.2. Diversity of the Tumour Immune Microenvironment
5. Use of Single-Cell Analysis to Identifying Biomarkers of Response to Immunotherapies and Novel Drug Targets
6. Future Perspectives in Incorporating Single-Cell Analysis into Clinical Trials and Routine Care of Cancer Patients
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, B.; Lin, Y.; Navin, N. Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell 2020, 37, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; van der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.C.; Braber, M.V.D.; Rozeman, E.A.; Haanen, J.B.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2020, 181, 747. [Google Scholar] [CrossRef]
- Murciano-Goroff, Y.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.; Harview, C.; Yearly, J.; Shintaku, I.; Taylor, E.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 168–571. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Sileni, V.C.; Gonzalez, R.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Schachter, J.; Long, G.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Hodi, F.S.; O'Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.; Cowey, C.L.; Dalle, S.; Schenker, M.; Sileni, V.C.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Eggermont, A.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.-J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final Version of 2009 AJCC Melanoma Staging and Classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Gide, T.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255.e6. [Google Scholar] [CrossRef] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013.e20. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Ma, Y.; Su, Z.; Liu, Y.; Liang, P.; Huang, C.; Liu, X.; Shao, J.; Zhang, Y.; Zhang, K.; et al. Single-cell RNA-sequencing analyses identify heterogeneity of CD8+ T cell subpopulations and novel therapy targets in melanoma. Mol. Ther. Oncolytics 2021, 20, 105–118. [Google Scholar] [CrossRef]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. 2017, 24, 1260–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorgulescu, J.B.; Braun, D.; Oliveira, G.; Keskin, D.B.; Wu, C.J. Acquired mechanisms of immune escape in cancer following immunotherapy. Genome Med. 2018, 10, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betters, D.M. Use of Flow Cytometry in Clinical Practice. J. Adv. Pract. Oncol. 2015, 6, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Zhang, W.; Xin, H.; Deng, G. Single Cell Isolation and Analysis. Front. Cell Dev. Biol. 2016, 4, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Wilmott, J.S.; Capper, D.; Preusser, M.; Zhang, Y.E.; Thompson, J.F.; Kefford, R.F.; von Deimling, A.; Scolyer, R.A. Immunohistochemistry Is Highly Sensitive and Specific for the Detection of V600E BRAF Mutation in Melanoma. Am. J. Surg. Pathol. 2013, 37, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Al-Kuraya, K.S. In-Situ Hybridization as a Molecular Tool in Cancer Diagnosis and Treatment. Curr. Med. Chem. 2012, 19, 3730–3738. [Google Scholar] [CrossRef]
- Giladi, A.; Amit, I. Single-Cell Genomics: A Stepping Stone for Future Immunology Discoveries. Cell 2018, 172, 14–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubin, M.M.; Esaulova, E.; Ward, J.P.; Malkova, O.N.; Runci, D.; Wong, P.; Noguchi, T.; Arthur, C.D.; Meng, W.; Alspach, E.; et al. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell 2018, 175, 1443. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.Z.; Roden, D.L.; Al-Eryani, G.; Bartonicek, N.; Harvey, K.; Cazet, A.S.; Chan, C.-L.; Junankar, S.; Hui, M.N.; Millar, E.A.; et al. Cryopreservation of human cancers conserves tumour heterogeneity for single-cell multi-omics analysis. Genome Med. 2021, 13, 1–17. [Google Scholar] [CrossRef]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar]
- Laehnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.; Hicks, S.; Robinson, M.D.; Vallejos, C.; Campbell, K.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 1–35. [Google Scholar] [CrossRef]
- Qi, R.; Ma, A.; Ma, Q.; Zou, Q. Clustering and classification methods for single-cell RNA-sequencing data. Brief. Bioinform. 2019, 21, 1196–1208. [Google Scholar] [CrossRef]
- Saelens, W.; Cannoodt, R.; Todorov, H.; Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 2019, 37, 547–554. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Andrews, T.S.; Hemberg, M. Identifying cell populations with scRNASeq. Mol. Asp. Med. 2018, 59, 114–122. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.; Wakimoto, H.; Cahill, D.; Nahed, B.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schelker, M.; Feau, S.; Du, J.; Ranu, N.; Klipp, E.; MacBeath, G.; Schoeberl, B.; Raue, A. Estimation of immune cell content in tumour tissue using single-cell RNA-seq data. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Dang, M.; Harada, K.; Han, G.; Wang, F.; Pizzi, M.P.; Zhao, M.; Tatlonghari, G.; Zhang, S.; Hao, D.; et al. Single-cell dissection of intratumoral heterogeneity and lineage diversity in metastatic gastric adenocarcinoma. Nat. Med. 2021, 27, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Cagnon, L.; Costa-Nunes, C.M.; Baumgaertner, P.; Montandon, N.; Leyvraz, L.; Michielin, O.; Romano, E.; Speiser, D.E. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 2013, 63, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglass, S.M.; Fane, M.E.; Sanseviero, E.; Ecker, B.L.; Kugel, C.H., 3rd; Behera, B.L.; Kumar, V.; Tcyganov, E.N.; Yin, X.; Liu, Q.; et al. Myeloid-Derived Suppressor Cells Are a Major Source of Wnt5A in the Melanoma Microenvironment and Depend on Wnt5A for Full Suppressive Activity. Cancer Res. 2021, 81, 658–670. [Google Scholar] [CrossRef]
- Pang, X.; Fan, H.-Y.; Tang, Y.-L.; Wang, S.-S.; Cao, M.-X.; Wang, H.-F.; Dai, L.-L.; Wang, K.; Yu, X.-H.; Wu, J.-B.; et al. Myeloid derived suppressor cells contribute to the malignant progression of oral squamous cell carcinoma. PLoS ONE 2020, 15, e0229089. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.; Rodman, C.; Su, M.-J.; Melms, J.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.-R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e24. [Google Scholar] [CrossRef] [Green Version]
- Reuben, A.; Spencer, C.N.; Prieto, P.A.; Gopalakrishnan, V.; Reddy, S.; Miller, J.P.; Mao, X.; De Macedo, M.P.; Chen, J.; Song, X.; et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom. Med. 2017, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Ko, M.E.; Cheng, H.; Zhu, R.; Xue, M.; Wang, J.; Lee, J.W.; Frankiw, L.; Xu, A.; Wong, S.; et al. Multi-omic single-cell snapshots reveal multiple independent trajectories to drug tolerance in a melanoma cell line. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Durante, M.A.; Rodriguez, D.A.; Kurtenbach, S.; Kuznetsov, J.N.; Sanchez, M.I.; Decatur, C.L.; Snyder, H.; Feun, L.G.; Livingstone, A.S.; Harbour, J.W. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.B.; Bordeaux, J.; Kim, J.Y.; Vaupel, C.; Rimm, D.L.; Ho, T.H.; Joseph, R.W.; Daud, A.I.; Conry, R.M.; Gaughan, E.M.; et al. Quantitative Spatial Profiling of PD-1/PD-L1 Interaction and HLA-DR/IDO-1 Predicts Improved Outcomes of Anti-PD-1 Therapies in Metastatic Melanoma. Clin. Cancer Res. 2018, 24, 5250–5260. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.; Efremova, M.; Riedel, A.; Mahata, B.; Pramanik, J.; Huuhtanen, J.; Kar, G.; Vento-Tormo, R.; Hagai, T.; Chen, X.; et al. Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Rep. 2020, 31, 107628. [Google Scholar] [CrossRef]
- Ennen, M.; Keime, C.; Kobi, D.; Mengus, G.; Lipsker, D.; Thibault-Carpentier, C.; Davidson, I. Single-cell gene expression signatures reveal melanoma cell heterogeneity. Oncogene 2014, 34, 3251–3263. [Google Scholar] [CrossRef]
- Ho, Y.-J.; Anaparthy, N.; Molik, D.; Mathew, G.; Aicher, T.; Patel, A.; Hicks, J.; Hammell, M.G. Single-cell RNA-seq analysis identifies markers of resistance to targeted BRAF inhibitors in melanoma cell populations. Genome Res. 2018, 28, 1353–1363. [Google Scholar] [CrossRef] [Green Version]
- Aissa, A.F.; Islam, A.B.M.M.K.; Ariss, M.M.; Go, C.C.; Rader, A.E.; Conrardy, R.D.; Gajda, A.M.; Rubio-Perez, C.; Valyi-Nagy, K.; Pasquinelli, M.; et al. Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer. Nat. Commun. 2021, 12, 1–25. [Google Scholar] [CrossRef]
- Mackay, L.K.; Minnich, M.; Kragten, N.A.M.; Liao, Y.; Nota, B.; Seillet, C.; Zaid, A.; Man, K.; Preston, S.; Freestone, D.; et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 2016, 352, 459–463. [Google Scholar] [CrossRef] [Green Version]
- Smalley, I.; Chen, Z.; Phadke, M.; Li, J.; Yu, X.; Wyatt, C.; Evernden, B.; Messina, J.L.; Sarnaik, A.; Sondak, V.K.; et al. Single-Cell Characterization of the Immune Microenvironment of Melanoma Brain and Leptomeningeal Metastases. Clin Cancer Res. 2021, 27. [Google Scholar] [CrossRef]
- Andreatta, M.; Corria-Osorio, J.; Müller, S.; Cubas, R.; Coukos, G.; Carmona, S.J. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat. Commun. 2021, 12, 1–19. [Google Scholar] [CrossRef]
- Nguyen, A.; Khoo, W.H.; Moran, I.; Croucher, P.; Phan, T.G. Single Cell RNA Sequencing of Rare Immune Cell Populations. Front. Immunol. 2018, 9, 1553. [Google Scholar] [CrossRef] [PubMed]
- Valihrach, L.; Androvic, P.; Kubista, M. Platforms for Single-Cell Collection and Analysis. Int. J. Mol. Sci. 2018, 19, 807. [Google Scholar] [CrossRef] [Green Version]
- Pellegrino, M.; Sciambi, A.; Yates, J.L.; Mast, J.D.; Silver, C.; Eastburn, D.J. RNA-Seq following PCR-based sorting reveals rare cell transcriptional signatures. BMC Genom. 2016, 17, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davey, K.; Wong, D.; Konopacki, F.; Kwa, E.; Ly, T.; Fiegler, H.; Sibley, C.R. A flexible microfluidic system for single-cell transcriptome profiling elucidates phased transcriptional regulators of cell cycle. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.D.; Chen, Y.-J.J.; Dunne, J.; Mir, A.; Hubschle, H.; Guillory, J.; Yuan, W.; Zhang, J.; Stinson, J.; Jaiswal, B.; et al. Massively parallel nanowell-based single-cell gene expression profiling. BMC Genom. 2017, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, D.; Boccalini, G.; Bonechi, M.; Biagioni, C.; Fassan, P.; Bertorelli, R.; De Sanctis, V.; Di Leo, A.; Migliaccio, I.; Malorni, L.; et al. ddSeeker: A tool for processing Bio-Rad ddSEQ single cell RNA-seq data. BMC Genom. 2018, 19, 960. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Zeisel, A.; Joost, S.; La Manno, G.; Zajac, P.; Kasper, M.; Lönnerberg, P.; Linnarsson, S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 2013, 11, 163–166. [Google Scholar] [CrossRef]
- Erickson, J.R.; Mair, F.; Bugos, G.; Martin, J.; Tyznik, A.J.; Nakamoto, M.; Mortimer, S.; Prlic, M. AbSeq Protocol Using the Nano-Well Cartridge-Based Rhapsody Platform to Generate Protein and Transcript Expression Data on the Single-Cell Level. STAR Protoc. 2020, 1, 100092. [Google Scholar] [CrossRef]
- Shum, E.Y.; Walczak, E.M.; Chang, C.; Fan, H.C. Quantitation of mRNA Transcripts and Proteins Using the BD Rhapsody™ Single-Cell Analysis System. Single Mol. Single Cell Seq. 2019, 1129, 63–79. [Google Scholar] [CrossRef]
- Han, X.; Zhou, Z.; Fei, L.; Sun, H.; Wang, R.; Chen, Y.; Chen, H.; Wang, J.; Tang, H.; Ge, W.; et al. Construction of a human cell landscape at single-cell level. Nature 2020, 581, 303–309. [Google Scholar] [CrossRef]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018, 173, 1307. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Laing, C.; Gao, J.; Burns, K.; Sarikonda, S.; Pollner, R.; Gong, H. Abstract 4290: Multi-omic single cell sequencing for deep cell immune profiling and identification of potential biomarkers for cell therapy and immunotherapy. Cancer Res. 2020, 80, 4290. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Zrimec, J.; Börlin, C.S.; Buric, F.; Muhammad, A.S.; Chen, R.; Siewers, V.; Verendel, V.; Nielsen, J.; Töpel, M.; Zelezniak, A. Deep learning suggests that gene expression is encoded in all parts of a co-evolving interacting gene regulatory structure. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Picelli, S.; Bjorklund, K.; Faridani, O.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Bjorklund, A.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 1–12. [Google Scholar] [CrossRef]
- See, P.; Lum, J.; Chen, J.; Ginhoux, F. A Single-Cell Sequencing Guide for Immunologists. Front. Immunol. 2018, 9, 2425. [Google Scholar] [CrossRef]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol. Cell 2017, 65, 631–643.e4. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Neff, N.F.; Kalisky, T.; Dalerba, P.; Treutlein, B.; Rothenberg, M.E.; Mburu, F.; Mantalas, G.L.; Sim, S.; Clarke, M.F.; et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods 2013, 11, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Svensson, V.; Natarajan, K.N.; Ly, L.-H.; Miragaia, R.J.; Labalette, C.; Macaulay, I.C.; Cvejic, A.; Teichmann, S.A. Power analysis of single-cell RNA-sequencing experiments. Nat. Methods 2017, 14, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Kashima, Y.; Sakamoto, Y.; Kaneko, K.; Seki, M.; Suzuki, Y.; Suzuki, A. Single-cell sequencing techniques from individual to multiomics analyses. Exp. Mol. Med. 2020, 52, 1419–1427. [Google Scholar] [CrossRef]
- Gao, C.; Zhang, M.; Chen, L. The Comparison of Two Single-cell Sequencing Platforms: BD Rhapsody and 10x Genomics Chromium. Curr. Genom. 2020, 21, 602–609. [Google Scholar] [CrossRef]
- Nichterwitz, S.; Chen, G.; Benitez, J.A.; Yilmaz, M.; Storvall, H.; Cao, M.; Sandberg, R.; Deng, Q.; Hedlund, E. Laser capture microscopy coupled with Smart-seq2 for precise spatial transcriptomic profiling. Nat. Commun. 2016, 7, 12139. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Allen, W.E.; Wright, M.A.; Sylwestrak, E.L.; Samusik, N.; Vesuna, S.; Evans, K.; Liu, C.; Ramakrishnan, C.; Liu, J.; et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 2018, 361, eaat5691. [Google Scholar] [CrossRef] [Green Version]
- Eng, C.-H.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.-C.; et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Ferrante, T.C.; Terry, R.; Turczyk, B.M.; Yang, J.L.; Lee, H.S.; Aach, J.; et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat. Protoc. 2015, 10, 442–458. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Yang, J.L.; Ferrante, T.C.; Terry, R.; Jeanty, S.S.F.; Li, C.; Amamoto, R.; et al. Highly Multiplexed Subcellular RNA Sequencing in Situ. Science 2014, 343, 1360–1363. [Google Scholar] [CrossRef] [Green Version]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Vickovic, S.; Eraslan, G.; Salmén, F.; Klughammer, J.; Stenbeck, L.; Schapiro, D.; Äijö, T.; Bonneau, R.; Bergenstråhle, L.; Navarro, J.F.; et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 2019, 16, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Bassiouni, R.; Gibbs, L.D.; Craig, D.W.; Carpten, J.D.; McEachron, T.A. Applicability of spatial transcriptional profiling to cancer research. Mol. Cell 2021, 81, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Bergenstråhle, J.; Lundeberg, J. Spatially Resolved Transcriptomes—Next Generation Tools for Tissue Exploration. BioEssays 2020, 42, 1900221. [Google Scholar] [CrossRef] [PubMed]
- Espina, V.; Wulfkuhle, J.D.; Calvert, V.S.; VanMeter, A.; Zhou, W.; Coukos, G.; Geho, D.H.; Petricoin, E.F., 3rd; Liotta, L.A. Laser-capture microdissection. Nat. Protoc. 2006, 1, 586–603. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Rozeman, E.A.; Fanchi, L.F.; Sikorska, K.; Van De Wiel, B.; Kvistborg, P.; Krijgsman, O.; Braber, M.V.D.; Philips, D.; Broeks, A.; et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat. Med. 2018, 24, 1655–1661. [Google Scholar] [CrossRef]
- Amaria, R.N.; Reddy, S.; Tawbi, H.A.; Davies, M.A.; Ross, M.I.; Glitza, I.C.; Cormier, J.N.; Lewis, C.; Hwu, W.-J.; Hanna, E.; et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 2018, 24, 1649–1654. [Google Scholar] [CrossRef]
- Waylen, L.N.; Nim, H.T.; Martelotto, L.G.; Ramialison, M. From whole-mount to single-cell spatial assessment of gene expression in 3D. Commun. Biol. 2020, 3, 1–11. [Google Scholar] [CrossRef]
- Marx, V. Method of the Year: Spatially resolved transcriptomics. Nat. Methods 2021, 18, 9–14. [Google Scholar] [CrossRef]
- Liao, J.; Lu, X.; Shao, X.; Zhu, L.; Fan, X. Uncovering an Organ’s Molecular Architecture at Single-Cell Resolution by Spatially Resolved Transcriptomics. Trends Biotechnol. 2020, 39, 43–58. [Google Scholar] [CrossRef]
- Vergara, I.A.; Mintoff, C.P.; Sandhu, S.; McIntosh, L.; Young, R.J.; Wong, S.Q.; Colebatch, A.; Cameron, D.L.; Kwon, J.L.; Wolfe, R.; et al. Evolution of late-stage metastatic melanoma is dominated by aneuploidy and whole genome doubling. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Larsen, M.S.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Schetters, S.T.T.; Rodriguez, E.; Kruijssen, L.J.W.; Crommentuijn, M.H.W.; Boon, L.; Bossche, J.V.D.; Haan, J.M.M.D.; Van Kooyk, Y. Monocyte-derived APCs are central to the response of PD1 checkpoint blockade and provide a therapeutic target for combination therapy. J. Immunother. Cancer 2020, 8, e000588. [Google Scholar] [CrossRef]
- Xiong, D.; Wang, Y.; You, M. A gene expression signature of TREM2hi macrophages and γδ T cells predicts immunotherapy response. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef]
- Thrane, K.; Eriksson, H.; Maaskola, J.; Hansson, J.; Lundeberg, J. Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. Cancer Res. 2018, 78, 5970–5979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [Green Version]
- Savas, P.; Kathleen Cuningham Foundation Consortium for research into Familial Breast cancer (kConFab); Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018, 24, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef] [PubMed]
- Hornburg, M.; Desbois, M.; Lu, S.; Guan, Y.; Lo, A.A.; Kaufman, S.; Elrod, A.; Lotstein, A.; DesRochers, T.M.; Munoz-Rodriguez, J.L.; et al. Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell 2021, 39, 928–944.e6. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal. Transduct. Target. Ther. 2021, 6, 1–12. [Google Scholar] [CrossRef]
- Fattore, L.; Ruggiero, C.F.; Liguoro, D.; Mancini, R.; Ciliberto, G. Single cell analysis to dissect molecular heterogeneity and disease evolution in metastatic melanoma. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Linch, M.; Goh, G.; Hiley, C.; Shanmugabavan, Y.; McGranahan, N.; Rowan, A.; Wong, Y.N.S.; King, H.; Furness, A.; Freeman, A.; et al. Intratumoural evolutionary landscape of high-risk prostate cancer: The PROGENY study of genomic and immune parameters. Ann. Oncol. 2017, 28, 2472–2480. [Google Scholar] [CrossRef]
- Rybinski, B.; Yun, K. Addressing intra-tumoral heterogeneity and therapy resistance. Oncotarget 2016, 7, 72322–72342. [Google Scholar] [CrossRef] [Green Version]
- Birkeland, E.; Zhang, S.; Poduval, D.; Geisler, J.; Nakken, S.; Vodak, D.; Meza-Zepeda, L.A.; Hovig, E.; Myklebost, O.; Knappskog, S.; et al. Patterns of genomic evolution in advanced melanoma. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Hayward, N.; Wilmott, J.; Waddell, N.; Johansson, P.A.; Field, M.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Rabbie, R.; Ansari-Pour, N.; Cast, O.; Lau, D.; Scott, F.; Welsh, S.J.; Parkinson, C.; Khoja, L.; Moore, L.; Tullett, M.; et al. Multi-site clonality analysis uncovers pervasive heterogeneity across melanoma metastases. Nat. Commun. 2020, 11, 4306. [Google Scholar] [CrossRef]
- Gonzalez-Silva, L.; Quevedo, L.; Varela, I. Tumor Functional Heterogeneity Unraveled by scRNA-seq Technologies. Trends Cancer 2020, 6, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Castagnoli, L.; Cancila, V.; Cordoba-Romero, S.L.; Faraci, S.; Talarico, G.; Belmonte, B.; Iorio, M.; Milani, M.; Volpari, T.; Chiodoni, C.; et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 2019, 38, 4047–4060. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, L.P.; Diaz, E.; Reya, T. The Role of the Microenvironment and Immune System in Regulating Stem Cell Fate in Cancer. Trends Cancer 2021, 7, 624–634. [Google Scholar] [CrossRef]
- McFarland, J.M.; Paolella, B.R.; Warren, A.; Geiger-Schuller, K.; Shibue, T.; Rothberg, M.; Kuksenko, O.; Colgan, W.N.; Jones, A.; Chambers, E.; et al. Multiplexed single-cell transcriptional response profiling to define cancer vulnerabilities and therapeutic mechanism of action. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Satas, G.; Zaccaria, S.; Mon, G.; Raphael, B.J. SCARLET: Single-Cell Tumor Phylogeny Inference with Copy-Number Constrained Mutation Losses. Cell Syst. 2020, 10, 323–332.e8. [Google Scholar] [CrossRef]
- Malikic, S.; Jahn, K.; Kuipers, J.; Sahinalp, S.C.; Beerenwinkel, N. Integrative inference of subclonal tumour evolution from single-cell and bulk sequencing data. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Ho, D.W.-H.; Tsui, Y.-M.; Chan, L.-K.; Sze, K.M.-F.; Zhang, X.; Cheu, J.W.-S.; Chiu, Y.-T.; Lee, J.M.-F.; Chan, A.C.-Y.; Cheung, E.T.-Y.; et al. Single-cell RNA sequencing shows the immunosuppressive landscape and tumor heterogeneity of HBV-associated hepatocellular carcinoma. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.F.; Wei, W.; Gupta, S.; Smithy, J.W.; Zelterman, D.; Kluger, H.M.; Rimm, D.L. Multiplex quantitative analysis of cancer-associated fibroblasts and immunotherapy outcome in metastatic melanoma. J. Immunother. Cancer 2019, 7, 194. [Google Scholar] [CrossRef] [Green Version]
- Rosati, E.; Dowds, C.M.; Liaskou, E.; Henriksen, E.K.K.; Karlsen, T.H.; Franke, A. Overview of methodologies for T-cell receptor repertoire analysis. BMC Biotechnol. 2017, 17, 61. [Google Scholar] [CrossRef]
- Han, J.; Zhao, Y.; Shirai, K.; Molodtsov, A.; Kolling, F.W.; Fisher, J.L.; Zhang, P.; Yan, S.; Searles, T.G.; Bader, J.M.; et al. Resident and circulating memory T cells persist for years in melanoma patients with durable responses to immunotherapy. Nat. Rev. Cancer 2021, 2, 300–311. [Google Scholar] [CrossRef]
- Griffiths, J.I.; Wallet, P.; Pflieger, L.T.; Stenehjem, D.; Liu, X.; Cosgrove, P.A.; Leggett, N.A.; McQuerry, J.A.; Shrestha, G.; Rossetti, M.; et al. Circulating immune cell phenotype dynamics reflect the strength of tumor–immune cell interactions in patients during immunotherapy. Proc. Natl. Acad. Sci. USA 2020, 117, 16072–16082. [Google Scholar] [CrossRef]
- Wu, T.D.; Madireddi, S.; de Almeida, P.E.; Banchereau, R.; Chen, Y.-J.J.; Chitre, A.S.; Chiang, E.Y.; Iftikhar, H.; O’Gorman, W.E.; Au-Yeung, A. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 2020, 579, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Delyon, J.; Mateus, C.; Lefeuvre, D.; Lanoy, E.; Zitvogel, L.; Chaput, N.; Roy, S.; Eggermont, A.M.M.; Routier, E.; Robert, C. Experience in daily practice with ipilimumab for the treatment of patients with metastatic melanoma: An early increase in lymphocyte and eosinophil counts is associated with improved survival. Ann. Oncol. 2013, 24, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Yoon, S.; Min, S.T.; Andersen, S.; Devitt, K.; Lam, P.Y.; Purdue, B.; Raghubar, A.; Hanson, S.J.; Jones, K.; et al. Spatial analysis of ligand-receptor interactions in skin cancer at genome-wide and single-cell resolution. bioRxiv 2021. [Google Scholar] [CrossRef]
- Swanton, C. Intratumor Heterogeneity: Evolution through Space and Time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef] [Green Version]
- Bedard, P.; Hansen, A.; Ratain, M.J.; Siu, L.L. Tumour heterogeneity in the clinic. Nature 2013, 501, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Cohen, Y.C.; Zada, M.; Wang, S.-Y.; Bornstein, C.; David, E.; Moshe, A.; Li, B.; Shlomi-Loubaton, S.; Gatt, M.E.; Gur, C.; et al. Identification of resistance pathways and therapeutic targets in relapsed multiple myeloma patients through single-cell sequencing. Nat. Med. 2021, 27, 491–503. [Google Scholar] [CrossRef] [PubMed]
| Key Findings | Single-Cell Platforms | Identified Cell Types | References |
|---|---|---|---|
| CD8 T cells associated with TCF7 transcription factor were predictive of immunotherapy response; exhausted T cells with abnormal activation of metabolic pathways are correlated with unfavourable prognosis | Smart-Seq2 | CD8+ T cell subtypes (exhausted, naïve and cytotoxic) | [16] |
| Dysfunctional CD8 T cells form a proliferative compartment within human melanoma; the abundance of dysfunctional T cells is associated with tumour recognition | MARS-Seq | Intratumoural CD4 and CD8 T cells | [4] |
| B cells and tertiary lymphoid structures promote ICB response and improve patient survival | Smart-Seq2 | B cells | [102,103] |
| Monocyte-derived APCs are central to the response of PD-1 checkpoint blockade and anti-CD40 is a potential novel treatment | Smart-Seq2 | Monocyte-derived dendritic cells | [104] |
| Macrophage and γδ T cell subtypes are overrepresented in non-responders to immunotherapy; gene expression signature of these innate cells can help predict treatment response. | Smart-Seq2 and 10x Genomics Chromium | TREM-high macrophages and γδ T cells | [105,106] |
| A cancer-associated transcriptional program promotes T cell exclusion and resistance to checkpoint immunotherapies | Smart-Seq2 | Melanoma cell (resistance signature associated with T cell exclusion and immune evasion) | [43] |
| Genetic heterogeneity in Stage III melanoma; coexistence of multiple melanoma signatures within a single tumour region | 10x Genomics Visium | Gene expression profiles of melanoma and lymphoid cells | [107] |
| Seven major subpopulations of CD8+ T cells are identified, of which, the exhausted T cell subpopulation is associated with unfavourable prognosis and increased in later-stage melanoma samples, while favourable naïve/memory and cytotoxic subpopulation cells are decreased | 10x Genomics Chromium | 7 representative subpopulations of CD8+ T cells | [17] |
| Cancer Type | Key Findings | Single-Cell Platforms | Identified Cell Types | References |
|---|---|---|---|---|
| Breast | Trajectory analysis on longitudinal samples demonstrated distinct T cell states associated with activation, hypoxia and terminal differentiation | 10x Genomics Chromium | CD45+ immune cells (Clusters of T cell, myeloid cell, B cell and NK cell) | [108] |
| Tumours with high TILs contained CD8+ T cells with features of TRM T cell differentiation and these CD8+ TRM cells expressed high levels of immune checkpoint molecules and effector proteins; CD8+ TRM gene signature significantly associated with improved patient survival | 10x Genomics Chromium | TREM-specific CD8+ T cells | [109] | |
| Cancer associated fibroblast clusters are linked to immunotherapy resistance, promote cancer cell differentiation and T cell exclusion | 10x Genomics Chromium | Cancer-associated fibroblast subsets | [110] | |
| Ovarian | Immune-desert tumours demonstrated low antigen presentation and enrichment of monocytes and immature macrophages; immune-infiltrated and -excluded tumours differ markedly in their T cell composition and fibroblast subsets; chemokine-receptor interactions were identified as potential mechanisms mediating immune cell infiltration | 10x Genomics Chromium | Tumour, stromal and immune cells | [111] |
| Lung | A high ratio of tumour-infiltrating “pre-exhausted” T cells to exhausted T cells was associated with better prognosis; a gene signature of activated tumour Tregs correlated with poor prognosis in lung adenocarcinoma | Smart-Seq2 | Peripheral blood, peritumoural and intratumoural T cells | [112] |
| Liver | Tumour-associated macrophages suppress T cell infiltration in hepatocellular carcinoma and TIGIT-NECTIN2 interaction regulates the immunosuppressive environment; transition of immune cells towards a more immunosuppressive and exhaustive status exemplifies the overall cancer-promoting immune landscape | 10x Genomics Chromium | Tumour and immune cells | [113] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quek, C.; Bai, X.; Long, G.V.; Scolyer, R.A.; Wilmott, J.S. High-Dimensional Single-Cell Transcriptomics in Melanoma and Cancer Immunotherapy. Genes 2021, 12, 1629. https://doi.org/10.3390/genes12101629
Quek C, Bai X, Long GV, Scolyer RA, Wilmott JS. High-Dimensional Single-Cell Transcriptomics in Melanoma and Cancer Immunotherapy. Genes. 2021; 12(10):1629. https://doi.org/10.3390/genes12101629
Chicago/Turabian StyleQuek, Camelia, Xinyu Bai, Georgina V. Long, Richard A. Scolyer, and James S. Wilmott. 2021. "High-Dimensional Single-Cell Transcriptomics in Melanoma and Cancer Immunotherapy" Genes 12, no. 10: 1629. https://doi.org/10.3390/genes12101629
APA StyleQuek, C., Bai, X., Long, G. V., Scolyer, R. A., & Wilmott, J. S. (2021). High-Dimensional Single-Cell Transcriptomics in Melanoma and Cancer Immunotherapy. Genes, 12(10), 1629. https://doi.org/10.3390/genes12101629