
Compound Heterozygote of Point Mutation and Chromosomal Microdeletion Involving OTUD6B Coinciding with ZMIZ1 Variant in Syndromic Intellectual Disability

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Cytogenomic Microarray
2.3. Whole Exome Sequencing Analysis
2.4. mRNA Study of ZMIZ1 Mutation
3. Results
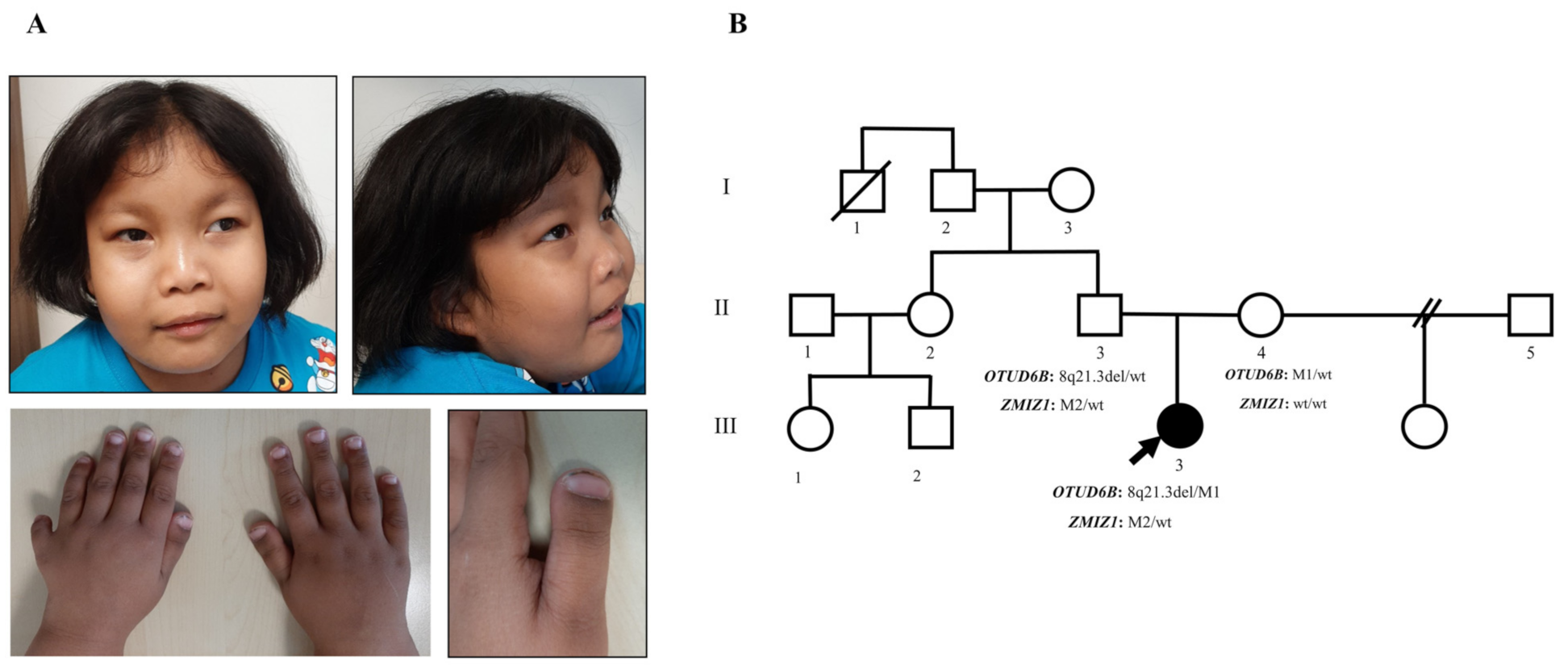
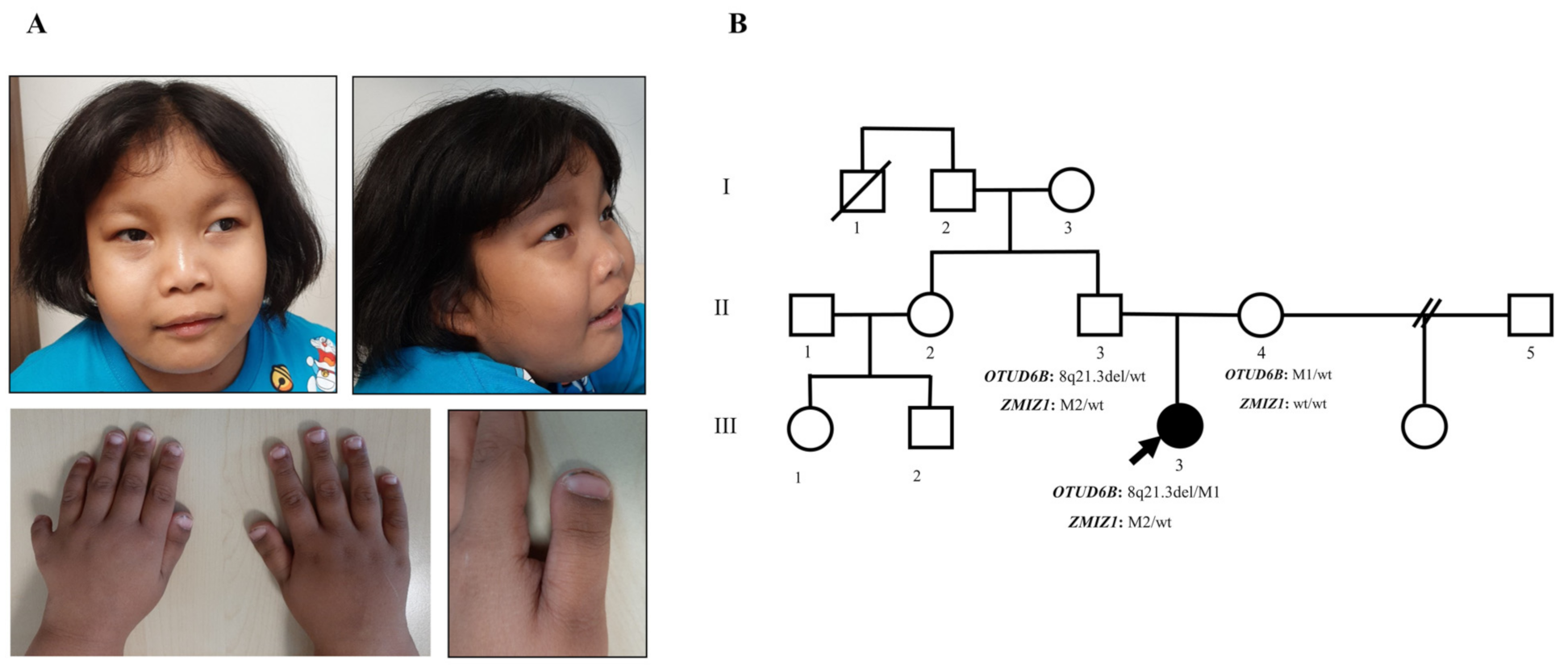
3.1. Clinical Manifestations and Family Data
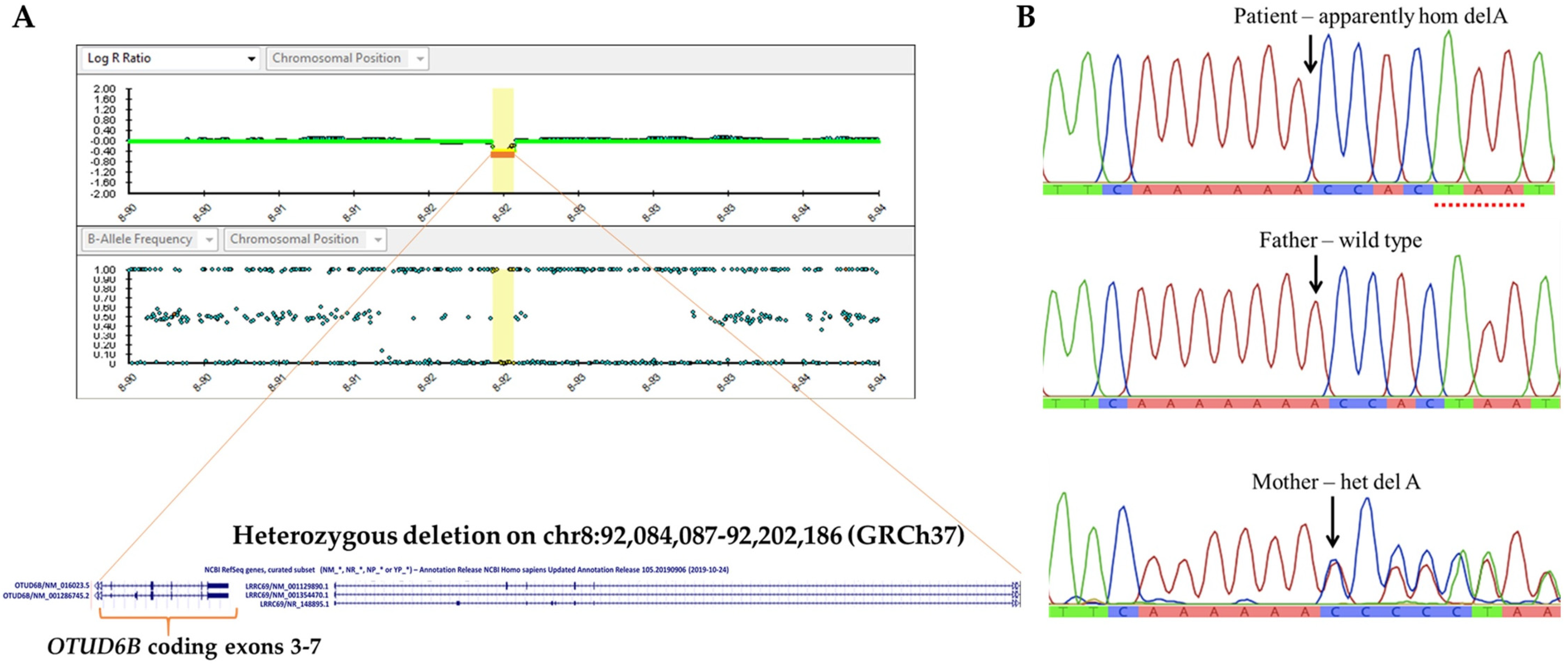
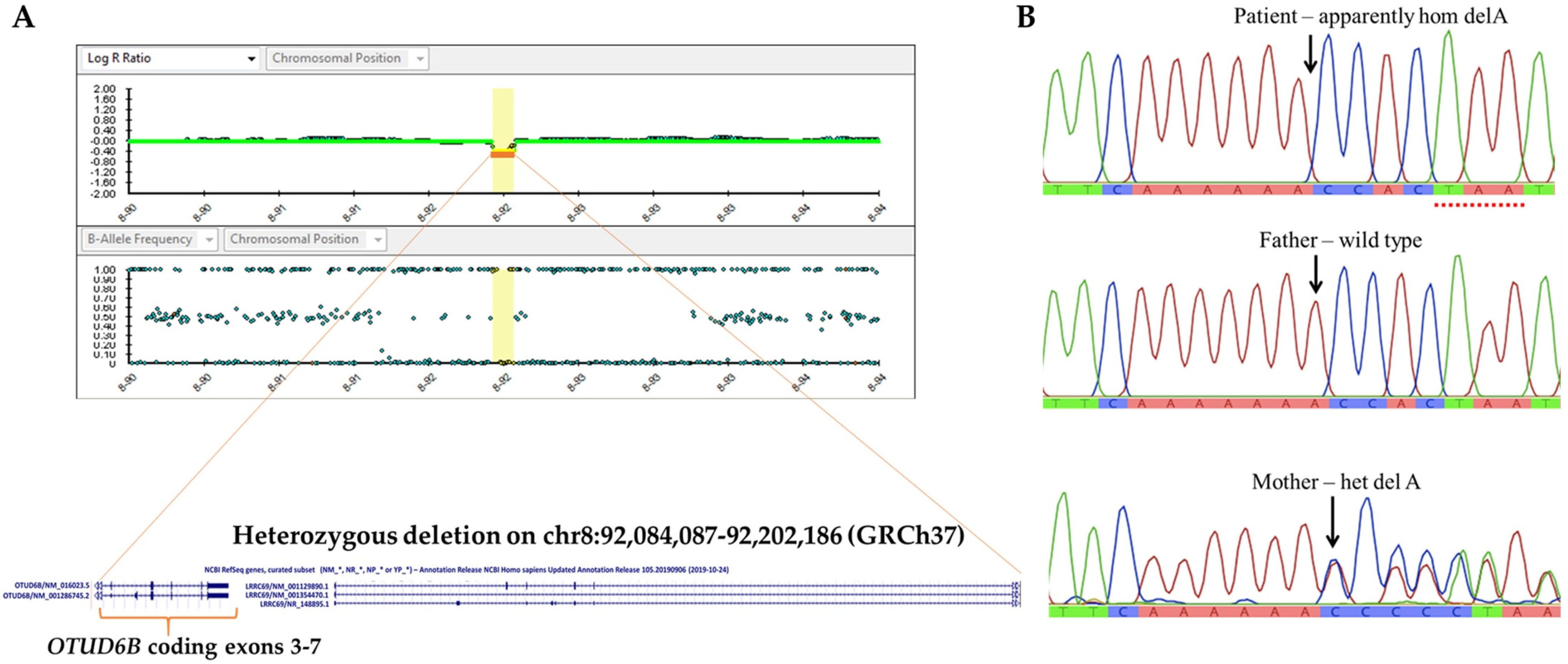
3.2. Identification of 8q21.3 Microdeletion Compounded with a Point Mutation Involving OTUD6B
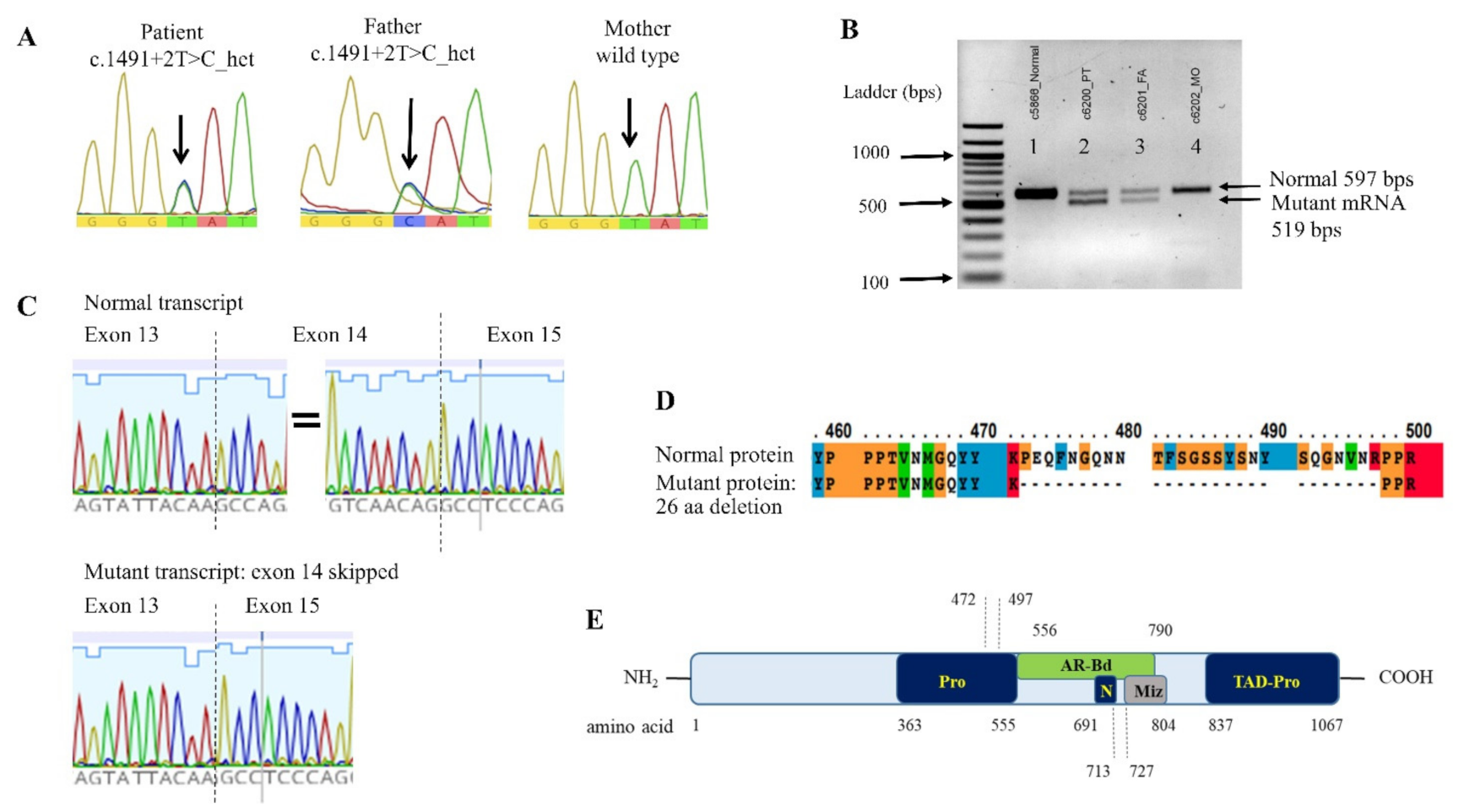
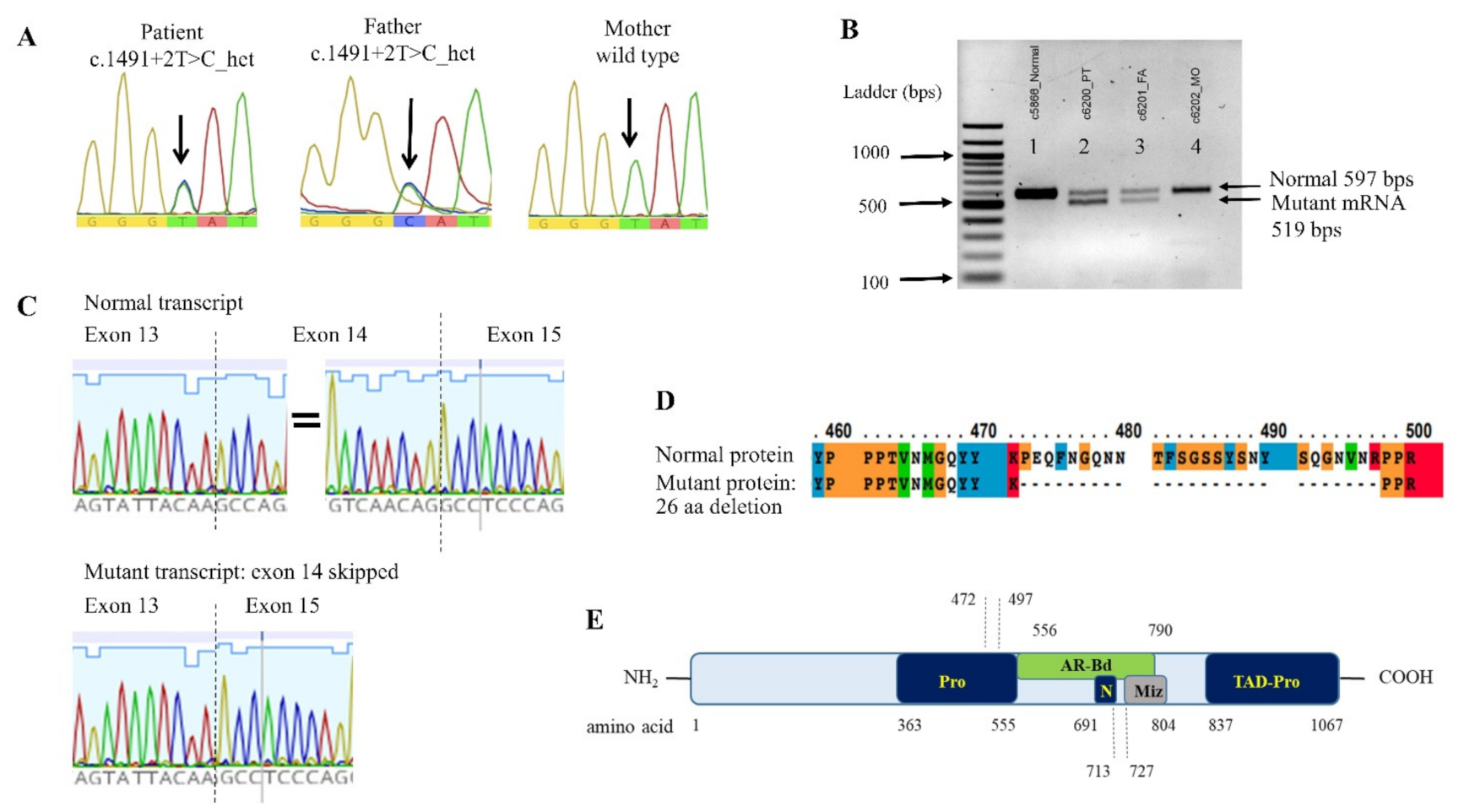
3.3. ZMIZ1 Splicing Mutation and its Effect on the mRNA Transcription
3.4. Clinical Similarities and Differences, Comparing between OTUD6B- and ZMIZ1-Related Phenotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Manning, M.; Hudgins, L. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet. Med. 2010, 12, 742–745. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Phung, Q.; Chan, S.; Chaudhari, R.; Quan, C.; O’Rourke, K.M.; Eby, M.; Pietras, E.; Cheng, G.; Bazan, J.F.; et al. DUBA: A deubiquitinase that regulates type I interferon production. Science 2007, 318, 1628–1632. [Google Scholar] [CrossRef] [PubMed]
- Sobol, A.; Askonas, C.; Alani, S.; Weber, M.J.; Ananthanarayanan, V.; Osipo, C.; Bocchetta, M. Deubiquitinase OTUD6B Isoforms Are Important Regulators of Growth and Proliferation. Mol. Cancer Res. 2017, 15, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago-Sim, T.; Burrage, L.C.; Ebstein, F.; Tokita, M.J.; Miller, M.; Bi, W.; Braxton, A.A.; Rosenfeld, J.A.; Shahrour, M.; Lehmann, A.; et al. Biallelic Variants in OTUD6B Cause an Intellectual Disability Syndrome Associated with Seizures and Dysmorphic Features. Am. J. Hum. Genet. 2017, 100, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Alkuraya, F.S. Phenotypic expansion of OTUD6B-related syndrome. Am. J. Med. Genet. Part A 2020, 182, 1530–1531. [Google Scholar] [CrossRef] [PubMed]
- Romero-Ibarguengoitia, M.E.; Cantú-Reyna, C.; Gutierrez-González, D.; Cruz-Camino, H.; González-Cantú, A.; Sanz Sánchez, M.A. Comparison of Genetic Variants and Manifestations of OTUD6B-Related Disorder: The First Mexican Case. J. Investig. Med. High Impact. Case Rep. 2020, 8, 2324709620957777. [Google Scholar] [CrossRef]
- Abdel-Salam, G.M.H.; Abdel-Hamid, M.S.; Sayed, I.S.M.; Zechner, U.; Bolz, H.J. OTUD6B-associated intellectual disability: Novel variants and genetic exclusion of retinal degeneration as part of a refined phenotype. J. Hum. Genet. 2021. [CrossRef]
- Sánchez-Soler, M.J.; Serrano-Antón, A.T.; López-González, V.; Ballesta Martínez, M.J.; Guillén-Navarro, E. First Spanish case of syndromic intellectual disability with dysmorphic facies, seizures, and distal limb anomalies caused by balletic mutations in the OTUD6B gene. Anal. Pediatr. 2020, 92, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Straniero, L.; Rimoldi, V.; Solda, G.; Bellini, M.; Biasucci, G.; Asselta, R.; Duga, S. First Replication of the Involvement of OTUD6B in Intellectual Disability Syndrome With Seizures and Dysmorphic Features. Front. Genet. 2018, 9, 464. [Google Scholar] [CrossRef] [Green Version]
- Carapito, R.; Ivanova, E.L.; Morlon, A.; Meng, L.; Molitor, A.; Erdmann, E.; Kieffer, B.; Pichot, A.; Naegely, L.; Kolmer, A.; et al. ZMIZ1 Variants Cause a Syndromic Neurodevelopmental Disorder. Am. J. Hum. Genet. 2020, 106, 137. [Google Scholar] [CrossRef] [Green Version]
- Latchman, K.; Calder, M.; Morel, D.; Rhodes, L.; Juusola, J.; Tekin, M. Autosomal dominant inheritance in a recently described ZMIZ1-related neurodevelopmental disorder: Case report of siblings and an affected parent. Am. J. Med. Genet. Part A 2020, 182, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Suktitipat, B.; Naktang, C.; Mhuantong, W.; Tularak, T.; Artiwet, P.; Pasomsap, E.; Jongjaroenprasert, W.; Fuchareon, S.; Mahasirimongkol, S.; Chantratita, W.; et al. Copy number variation in Thai population. PLoS ONE 2014, 9, e104355. [Google Scholar] [CrossRef]
- Seo, G.H.; Kim, T.; Choi, I.H.; Park, J.Y.; Lee, J.; Kim, S.; Won, D.G.; Oh, A.; Lee, Y.; Choi, J.; et al. Diagnostic yield and clinical utility of whole exome sequencing using an automated variant prioritization system, EVIDENCE. Clin. Genet. 2020, 98, 562–570. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Greene, D.; Richardson, S.; Turro, E. Phenotype Similarity Regression for Identifying the Genetic Determinants of Rare Diseases. Am. J. Hum. Genet. 2016, 98, 490–499. [Google Scholar] [CrossRef] [Green Version]
- Köhler, S.; Schulz, M.H.; Krawitz, P.; Bauer, S.; Dölken, S.; Ott, C.E.; Mundlos, C.; Horn, D.; Mundlos, S.; Robinson, P.N. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am. J. Hum. Genet. 2009, 85, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Iafrate, A.J.; Brothman, A.R. Copy number variations and clinical cytogenetic diagnosis of constitutional disorders. Nat. Genet. 2007, 39, S48–S54. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Kasher, M.; Malkina, I.; Livshits, G. Is craniofacial morphology and body composition related by common genes: Comparative analysis of two ethnically diverse populations. Am. J. Phys. Anthropol. 2021, 176, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Lumaka, A.; Cosemans, N.; Lulebo Mampasi, A.; Mubungu, G.; Mvuama, N.; Lubala, T.; Mbuyi-Musanzayi, S.; Breckpot, J.; Holvoet, M.; de Ravel, T.; et al. Facial dysmorphism is influenced by ethnic background of the patient and of the evaluator. Clin. Genet. 2017, 92, 166–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuai, K.; Liu, B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Gaiano, N.; Nye, J.S.; Fishell, G. Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron 2000, 26, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Angulo-Rojo, C.; Manning-Cela, R.; Aguirre, A.; Ortega, A.; López-Bayghen, E. Involvement of the Notch pathway in terminal astrocytic differentiation: Role of PKA. ASN Neuro 2013, 5, e00130. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | This Study (n = 1) | OTUD6B | ZMIZ1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Santiago-Sim et al. [5] | Straniero et al. [10] | Sánchez-Soler et al. [9] | Alkuraya et al. [6] | Romero-Ibarguengoitia et al. [7] | Abdel-Salam et al. [8] | Frequency | Carapito et al. [11] | Latchmanet al. [12] | Frequency | ||
| (n = 12) | (n = 1) | (n = 1) | (n = 1) | (n = 1) | (n = 5) | (n = 21) | (n = 19) | (n = 3) | (n = 22) | ||
| Central nervous system | |||||||||||
| Intellectual disability | + | 12 | + | + | NA | + | 5/5 | 20/20 (100%) | 19 | 3/3 | 22/23 (100%) |
| Epilepsy | + | 12 | + | + | ++ | + | 3/4 | 19/20 (95%) | 3/18 | − | 3/18 (16.7%) |
| Hypotonia | − | 9 | + | + | NA | + | NA | 12/15 (80%) | 10/16 | 2/3 | 12/19 (63.2%) |
| Motor delay | + | 9 | + | NA | + | + | 5/5 | 17/20 (85%) | 12/16 | 2/2 | 14/18 (77.8%) |
| Speech delay | + | 9 | + | + | NA | + | 3/5 | 15/20 (75%) | 15/17 | 3/3 | 18/20 (90%) |
| CNS anomalies | − | 6 | − | + | NA | + | 3/4 | 11/19 (57.9%) | 7/14 | − | 7/17 (41.2%) |
| Microcephaly | − | 4 | + | + | + | + | 4/4 | 12/20 (60%) | 8/10 | 1/3 | 9/13 (69.2%) |
| Facial dysmorphism | |||||||||||
| Ptosis | − | 1 | + | + | + | − | 2/5 | 6/21 (28.6%) | 3/17 | 3/3 | 6/20 (30%) |
| Downslant pf. | − | 2 | − | − | − | − | − | 2/21 (9.5%) | 2/18 | 1/3 | 3/21 (14.3%) |
| Long pf. | − | 6 | − | + | NA | + | NA | 8/15 (53.3%) | 1/19 | − | 1/22 (4.5%) |
| Arched eyebrows | − | 3 | − | + | + | + | NA | 6/16 (37.5%) | 1/19 | − | 1/22 (4.5%) |
| Prominent nasal bridge | + | 5 | − | − | + | + | NA | 7/16 (43.8%) | 2/19 | − | 2/22 (9.1%) |
| Long philtrum | + | 6 | + | + | + | + | 4/5 | 14/21 (66.7%) | 2/19 | − | 2/22 (9.1%) |
| Thin vermillion of upper lip | + | 5 | + | + | + | + | 1/5 | 10/21 (47.6%) | − | 1/3 | 1/22 (4.5%) |
| Micro/retrognathia | − | 2 | − | NA | + | − | − | 3/20 (15%) | 3/19 | 2/3 | 5/22 (22.7%) |
| Ear anomalies | − | 7 | + | + | + | + | 3/5 | 14/21 (66.7%) | 4/19 | 1/3 | 5/22 (22.7%) |
| Congenital heart disease | + | 4 | + | − | + | + | 1/3 | 8/19 (42.1%) | 4/17 | 2/3 | 6/20 (30%) |
| Gastrointestinal system | |||||||||||
| Feeding difficulties | + | 9 | + | + | + | + | NA | 13/16 (81.3%) | 9/17 | 2/2 | 11/19 (57.9%) |
| Constipation | − | 2 | NA | + | NA | + | NA | 4/14 (28.6%) | NA | NA | NA |
| Skeletal system | |||||||||||
| Distal limb anomalies | |||||||||||
| Broad thumbs | + | 6 | + | + | + | − | 5/5 | 14/21 (66.7%) | − | − | − |
| Polydactyly | + | − | − | − | + | + | − | 2/21 (9.5%) | − | − | − |
| Others a | − | 9 | − | + | − | − | − | 10/21 (47.6%) | 12/18 | - | 12/21 (57.1%) |
| Sacral dimple | − | 2 | − | − | NA | + | 1/5 | 4/20 (20%)) | − | − | − |
| Scoliosis | − | 5 | − | − | − | + | NA | 6/16 (37.5%) | − | − | − |
| Endocrine system | |||||||||||
| Hypothyroidism | − | 2 | NA | NA | NA | + | − | 3/18 (16.7%) | NA | NA | NA |
| Opthalmologic abnormalities | − | NA | + b | + c | − | − | (2/5) d | 2/9 (22.2%) | 10/17 e | 1/3 f | 11/20 (55%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phetthong, T.; Khongkrapan, A.; Jinawath, N.; Seo, G.-H.; Wattanasirichaigoon, D. Compound Heterozygote of Point Mutation and Chromosomal Microdeletion Involving OTUD6B Coinciding with ZMIZ1 Variant in Syndromic Intellectual Disability. Genes 2021, 12, 1583. https://doi.org/10.3390/genes12101583
Phetthong T, Khongkrapan A, Jinawath N, Seo G-H, Wattanasirichaigoon D. Compound Heterozygote of Point Mutation and Chromosomal Microdeletion Involving OTUD6B Coinciding with ZMIZ1 Variant in Syndromic Intellectual Disability. Genes. 2021; 12(10):1583. https://doi.org/10.3390/genes12101583
Chicago/Turabian StylePhetthong, Tim, Arthaporn Khongkrapan, Natini Jinawath, Go-Hun Seo, and Duangrurdee Wattanasirichaigoon. 2021. "Compound Heterozygote of Point Mutation and Chromosomal Microdeletion Involving OTUD6B Coinciding with ZMIZ1 Variant in Syndromic Intellectual Disability" Genes 12, no. 10: 1583. https://doi.org/10.3390/genes12101583
APA StylePhetthong, T., Khongkrapan, A., Jinawath, N., Seo, G.-H., & Wattanasirichaigoon, D. (2021). Compound Heterozygote of Point Mutation and Chromosomal Microdeletion Involving OTUD6B Coinciding with ZMIZ1 Variant in Syndromic Intellectual Disability. Genes, 12(10), 1583. https://doi.org/10.3390/genes12101583