
Uptake and Patient Perspectives on Additional Testing for Novel Disease-Associated Genes: Lessons from a PAH Cohort

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
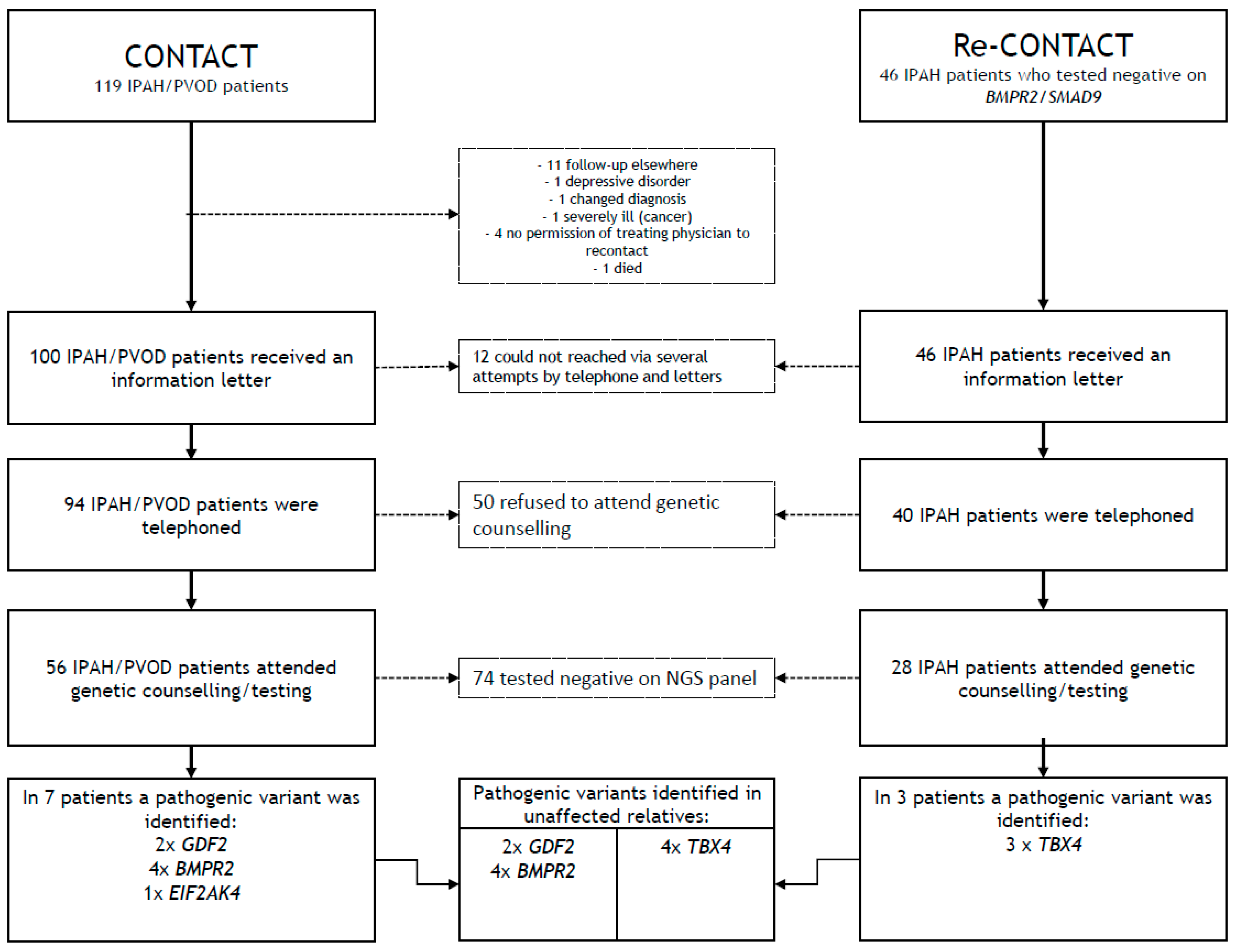
2. (Re-)Contact Procedure
2.1. Methods
2.2. Results
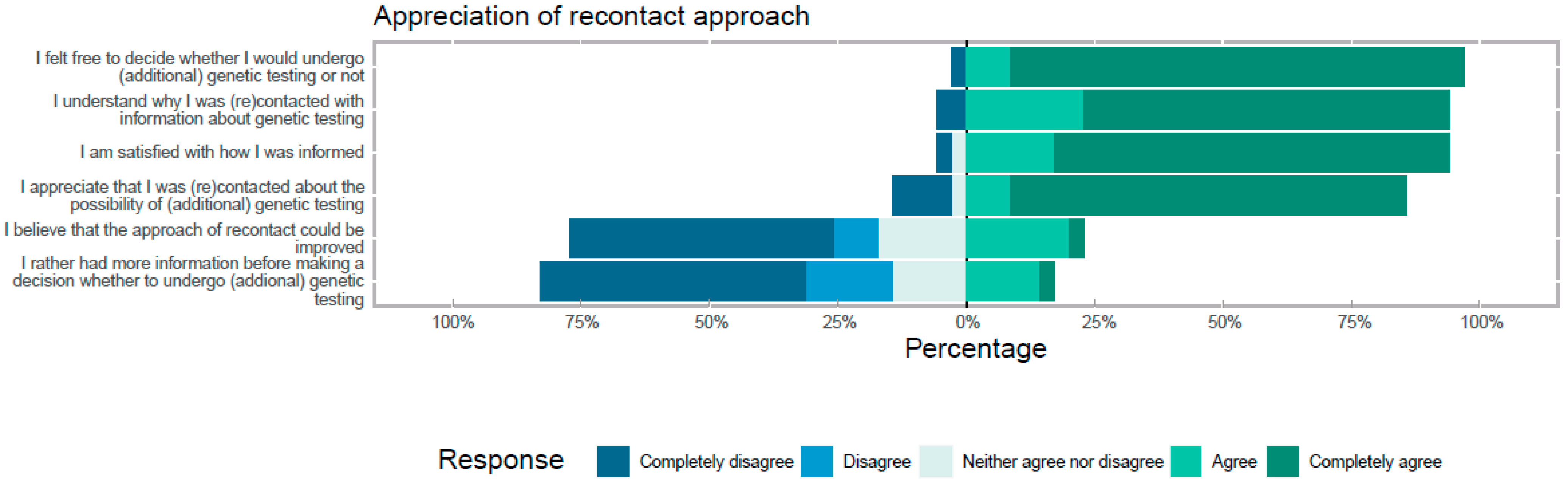
3. Patient Perspectives
3.1. Methods
3.2. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galiè, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [PubMed]
- Ling, Y.; Johnson, M.K.; Kiely, D.G.; Condliffe, R.; Elliot, C.A.; Gibbs, J.S.R.; Howard, L.S.; Pepke-Zaba, J.; Sheares, K.K.K.; Corris, P.A.; et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: Results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am. J. Respir. Crit. Care Med. 2012, 186, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chung, W.K. The role of genetics in pulmonary arterial hypertension. J. Pathol. 2017, 241, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadinnapola, C.; Bleda, M.; Haimel, M.; Screaton, N.; Swift, A.; Dorfmüller, P.; Preston, S.D.; Southwood, M.; Hernandez-Sanchez, J.; Martin, J.; et al. Phenotypic Characterization of EIF2AK4 Mutation Carriers in a Large Cohort of Patients Diagnosed Clinically With Pulmonary Arterial Hypertension. Circulation 2017, 136, 2022–2033. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zeng, Q.; Ma, Y.; Liu, B.; Chen, Q.; Li, W.; Xiong, C.; Zhou, Z. Genetic analyses in a cohort of 191 pulmonary arterial hypertension patients. Respir. Res. 2018, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- van den Heuvel, L.M.; Jansen, S.; Alsters, S.I.; Post, M.C.; van der Smagt, J.J.; Man, H.D.; van Tintelen, J.P.; Gille, H.; Christiaans, I.; Noordegraaf, A.V.; et al. Genetic Evaluation in a Cohort of 126 Dutch Pulmonary Arterial Hypertension Patients. Genes 2020, 11, 1191. [Google Scholar] [CrossRef] [PubMed]
- Gräf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, R.D.; Aldred, M.A.; James, V.; Harrison, R.E.; Patel, B.; Schwalbe, E.C.; Gruenig, E.; Janssen, B.; Koehler, R.; Seeger, W.; et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum. Mutat. 2006, 27, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Montani, D.; Lau, E.M.; Dorfmüller, P.; Girerd, B.; Jaïs, X.; Savale, L.; Perros, F.; Nossent, E.; Garcia, G.; Parent, F.; et al. Pulmonary veno-occlusive disease. Eur. Respir. J. 2016, 47, 1518–1534. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M.; Gerry Coghlan, J.; Khanna, D. Early detection and management of pulmonary arterial hypertension. Eur. Respir. Rev. 2012, 21, 306–312. [Google Scholar] [CrossRef]
- Montani, D.; Girerd, B.; Jaïs, X.; Laveneziana, P.; Lau, E.M.; Bouchachi, A.; Hascoët, S.; Günther, S.; Godinas, L.; Parent, F.; et al. Screening for pulmonary arterial hypertension in adults carrying a BMPR2 mutation. Eur. Respir. J. 2021, 58, 2004229. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Spinhoven, P.; Ormel, J.; Sloekers, P.P.; Kempen, G.I.; Speckens, A.E.; Van Hemert, A.M. A validation study of the Hospital Anxiety and Depression Scale (HADS) in different groups of Dutch subjects. Psychol. Med. 1997, 27, 363–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douma, K.F.; Aaronson, N.K.; Vasen, H.F.; Gerritsma, M.A.; Gundy, C.M.; Janssen, E.P.A.; Vriends, A.H.J.T.; Cats, A.; Verhoef, S.; Bleiker, E.M.A. Psychological distress and use of psychosocial support in familial adenomatous polyposis. Psychooncology. 2010, 19, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, D.; Lucassen, A.M.; Clarke, A.; Dheensa, S.; Doheny, S.; Turnpenny, P.D.; Kelly, S.E. Recontact in clinical practice: A survey of clinical genetics services in the United Kingdom. Genet. Med. 2016, 18, 876–881. [Google Scholar] [CrossRef] [Green Version]
- Giesbertz, N.A.A.; van Harten, W.H.; Bredenoord, A.L. A duty to recontact in genetics: Context matters. Nat. Rev. Genet. 2019, 20, 371–372. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jansen, S.M.A.; van de Heuvel, L.M.; Houweling, A.C.; van Tintelen, J.P.; de Man, F.S.; Vonk Noordegraaf, A.; Jan Bogaard, H. Uptake and Patient Perspectives on Additional Testing for Novel Disease-Associated Genes: Lessons from a PAH Cohort. Genes 2021, 12, 1540. https://doi.org/10.3390/genes12101540
Jansen SMA, van de Heuvel LM, Houweling AC, van Tintelen JP, de Man FS, Vonk Noordegraaf A, Jan Bogaard H. Uptake and Patient Perspectives on Additional Testing for Novel Disease-Associated Genes: Lessons from a PAH Cohort. Genes. 2021; 12(10):1540. https://doi.org/10.3390/genes12101540
Chicago/Turabian StyleJansen, Samara M. A., Lieke M. van de Heuvel, Arjan C. Houweling, J. Peter van Tintelen, Frances S. de Man, Anton Vonk Noordegraaf, and Harm Jan Bogaard. 2021. "Uptake and Patient Perspectives on Additional Testing for Novel Disease-Associated Genes: Lessons from a PAH Cohort" Genes 12, no. 10: 1540. https://doi.org/10.3390/genes12101540
APA StyleJansen, S. M. A., van de Heuvel, L. M., Houweling, A. C., van Tintelen, J. P., de Man, F. S., Vonk Noordegraaf, A., & Jan Bogaard, H. (2021). Uptake and Patient Perspectives on Additional Testing for Novel Disease-Associated Genes: Lessons from a PAH Cohort. Genes, 12(10), 1540. https://doi.org/10.3390/genes12101540