
MIA3 Splice Defect in Cane Corso Dogs with Dental-Skeletal-Retinal Anomaly (DSRA)

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical and Pathological Examinations
2.2. DNA and SNV Genotyping
2.3. Linkage Analysis and Homozygosity Mapping
2.4. Whole-Genome Sequencing
2.5. Variant Calling
2.6. Gene Analysis
2.7. Sanger Sequencing
2.8. RNA Isolation and RT-PCR
3. Results
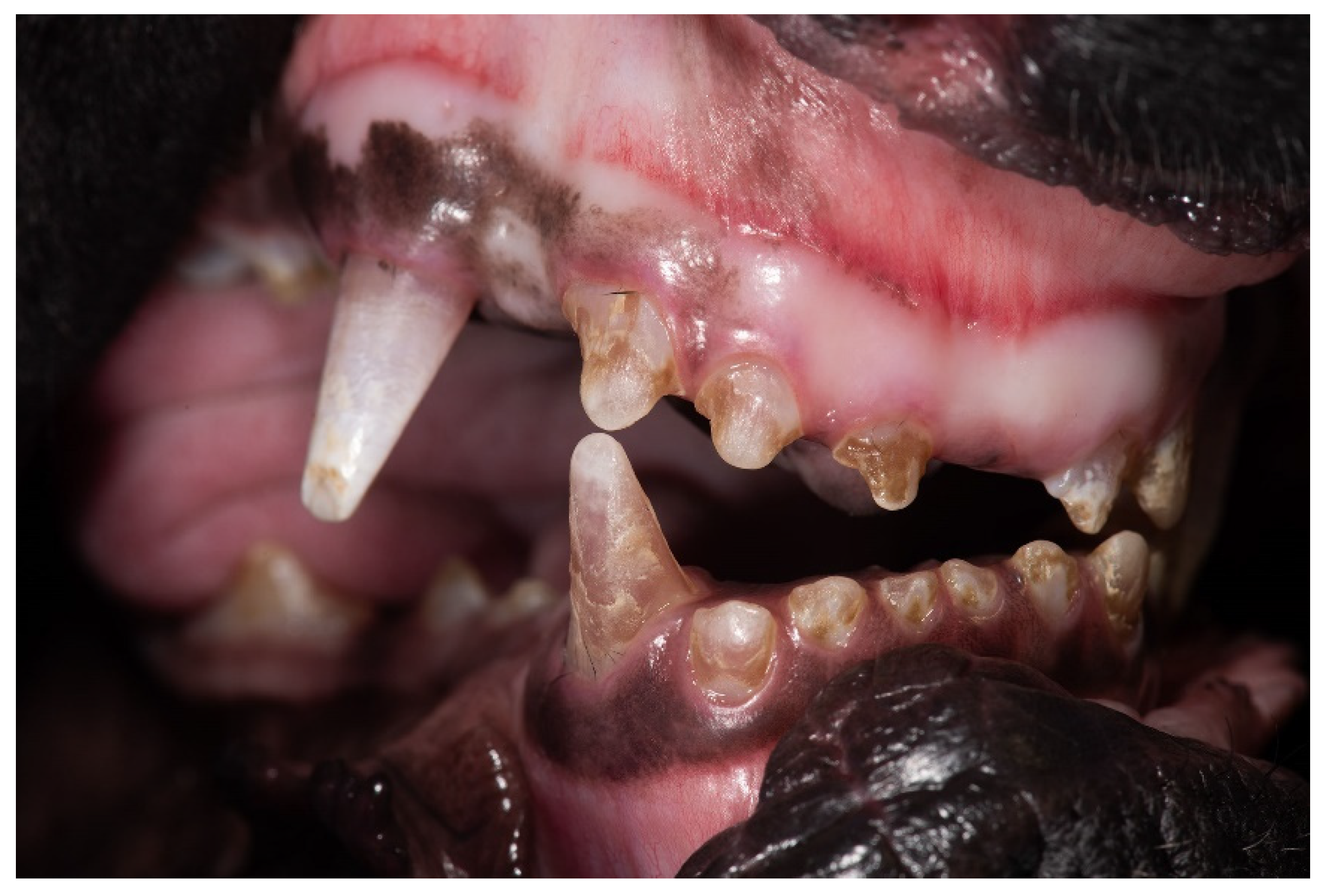
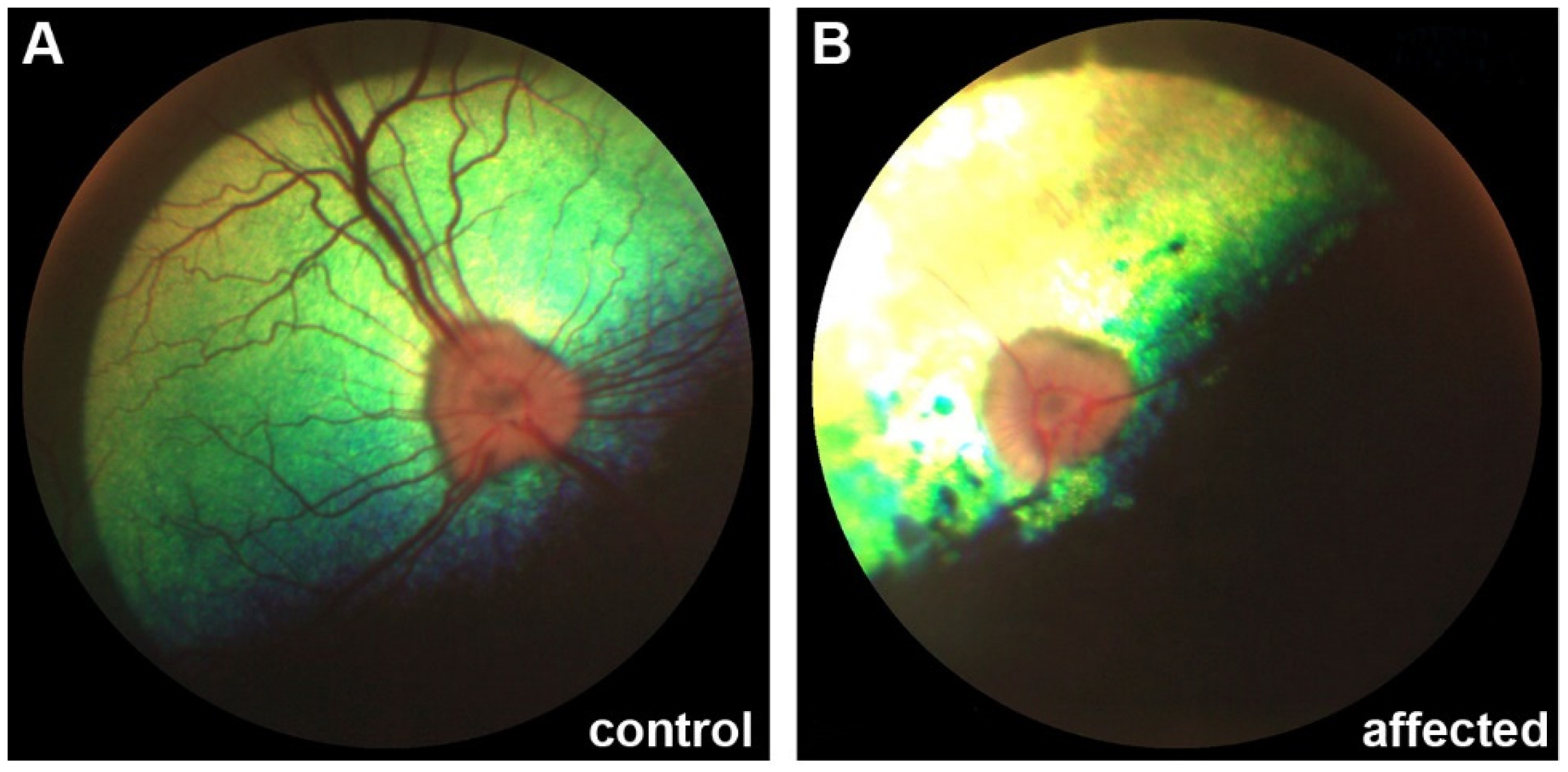
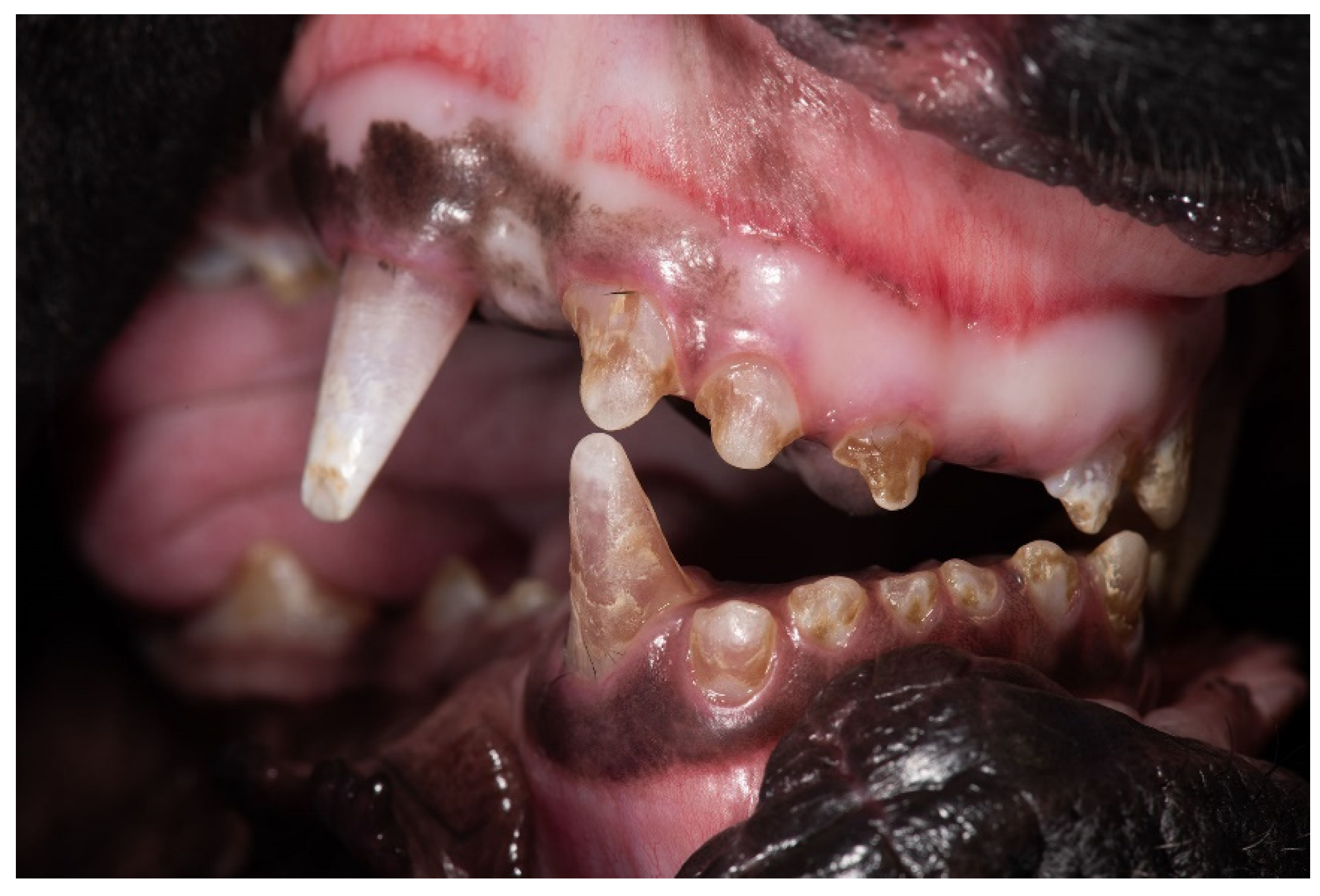
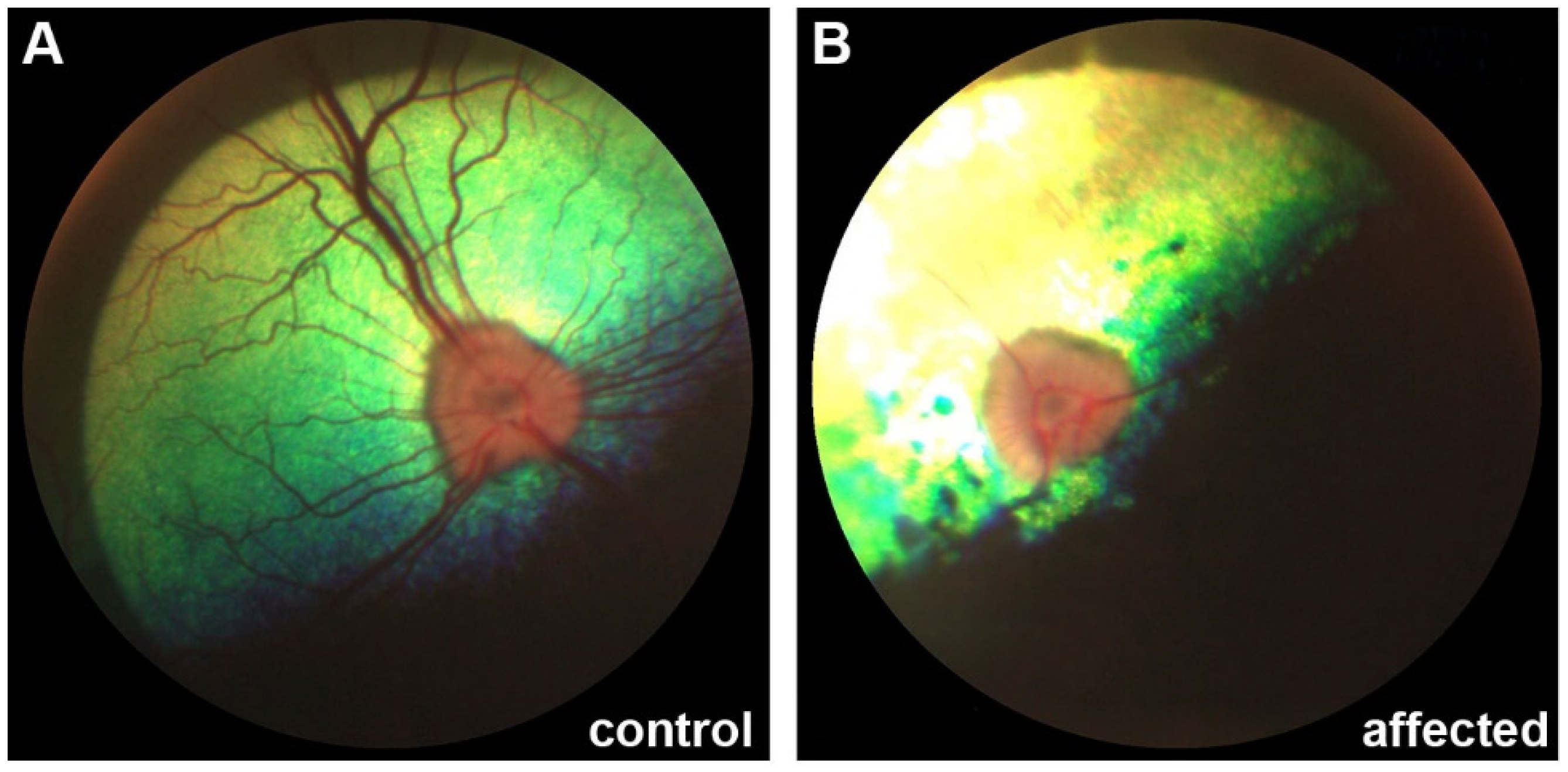
3.1. Clinical Description
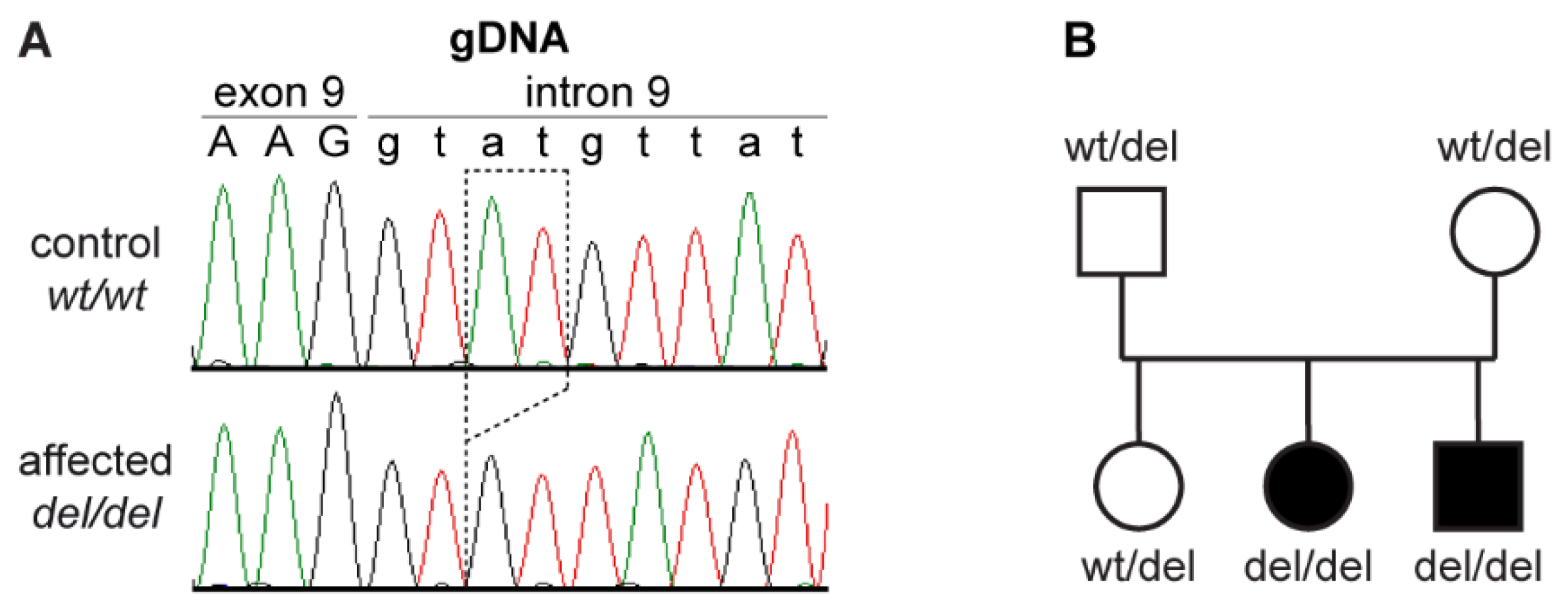
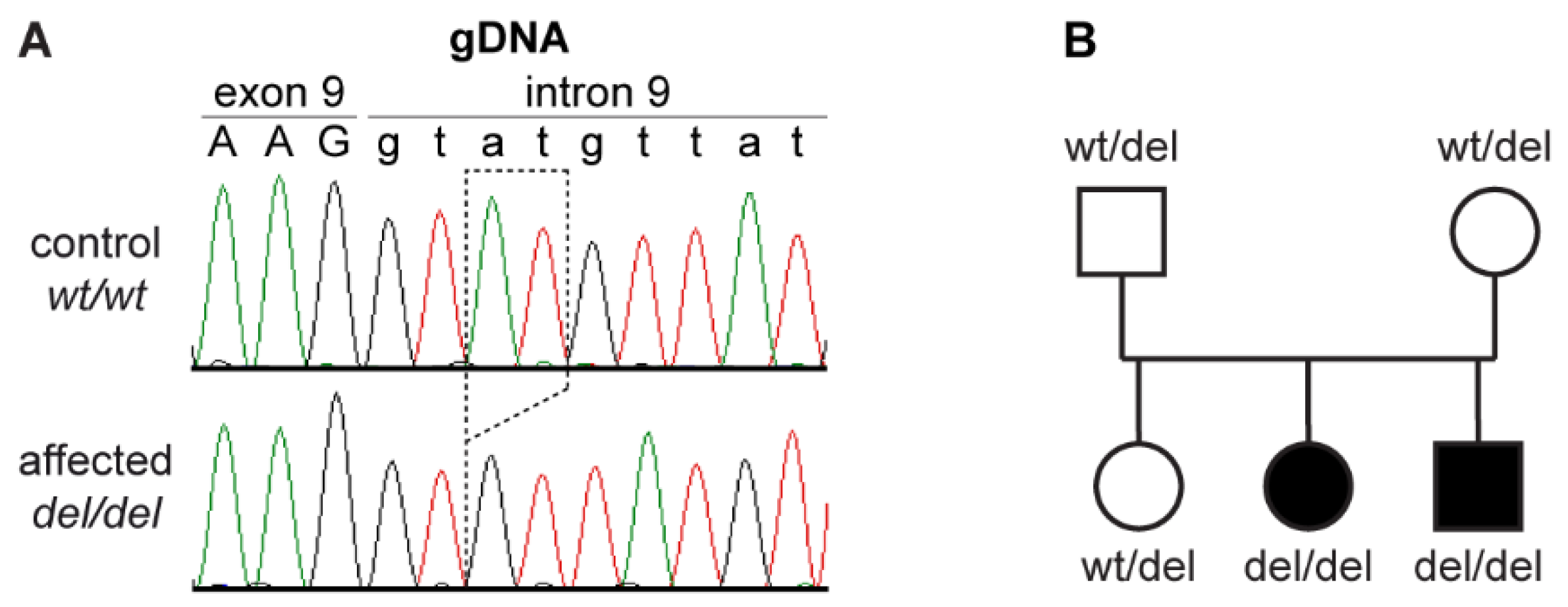
3.2. Genetic Analysis
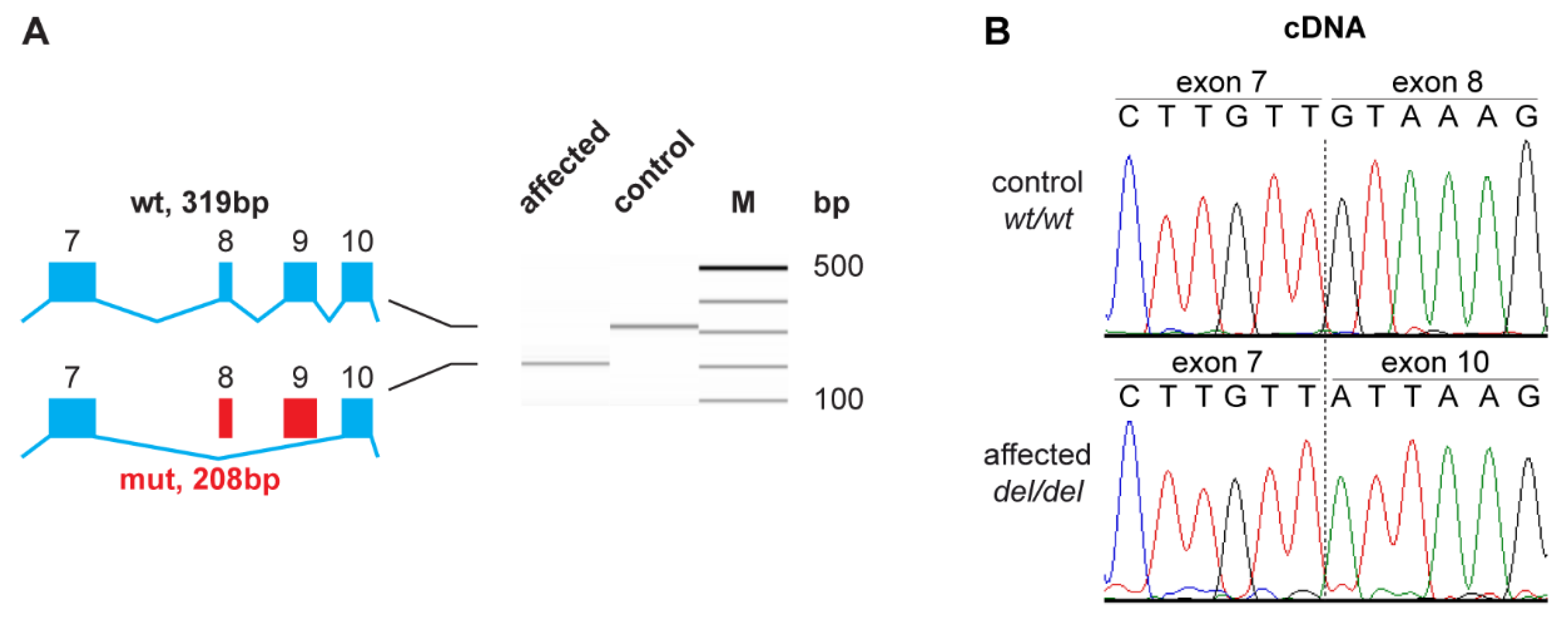
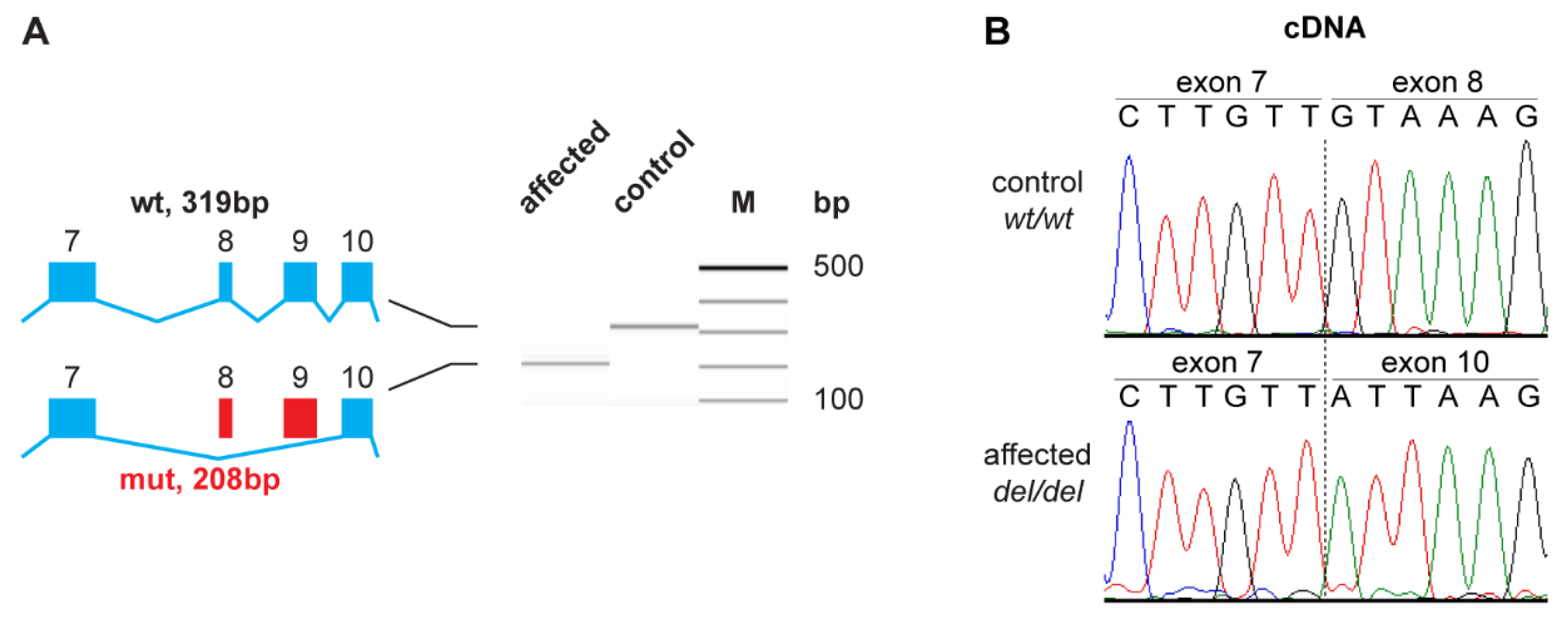
3.3. Functional Confirmation at the Transcript Level
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krakow, D.; Rimoin, D.L. The skeletal dysplasias. Genet. Med. 2010, 12, 327–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. Part A 2019, 179, 2393–2419. [Google Scholar] [CrossRef] [PubMed]
- Carrig, C.B.; MacMillan, A.; Brundage, S.; Pool, R.R.; Morgan, J.P. Retinal dysplasia associated with skeletal abnormalities in Labrador Retrievers. J. Am. Vet. Med. Assoc. 1977, 170, 49–57. [Google Scholar]
- Carrig, C.B.; Sponenberg, D.P.; Schmidt, G.M.; Tvedten, H.W. Inheritance of associated ocular and skeletal dysplasia in Labrador retrievers. J. Am. Vet. Med. Assoc. 1988, 193, 1269–1272. [Google Scholar]
- Meyers, V.N.; Jezyk, P.F.; Aguirre, G.D.; Patterson, D.F. Short-limbed dwarfism and ocular defects in the Samoyed dog. J. Am. Vet. Med. Assoc. 1983, 183, 975–979. [Google Scholar]
- Goldstein, O.; Guyon, R.; Kukekova, A.; Kuznetsova, T.N.; Pearce-Kelling, S.E.; Johnson, J.; Aguirre, G.D.; Acland, G.M. COL9A2 and COL9A3 mutations in canine autosomal recessive oculoskeletal dysplasia. Mamm. Genome 2010, 21, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Stavinohova, R.; Hartley, C.; Burmeister, L.M.; Ricketts, S.L.; Pettitt, L.; Tetas Pont, R.; Hitti, R.J.; Schofield, E.; Oliver, J.A.C.; Mellersh, C.S. Clinical, histopathological and genetic characterisation of oculoskeletal dysplasia in the Northern Inuit Dog. PLoS ONE 2019, 14, e0220761. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Abecasis, G.R.; Cherny, S.S.; Cookson, W.O.; Cardon, L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002, 30, 97–101. [Google Scholar] [CrossRef]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Dog Biomedical Variant Database Consortium (DBVDC). A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Christen, M.; Austel, M.; Banovic, F.; Jagannathan, V.; Leeb, T. NSDHL Frameshift Deletion in a Mixed Breed Dog with Progressive Epidermal Nevi. Genes 2020, 11, 1297. [Google Scholar] [CrossRef]
- Sebbag, L.; Riggs, A.; Carnevale, J. Oculo-skeletal dysplasia in five Labrador Retrievers. Vet. Ophthalmol. 2020, 23, 386–393. [Google Scholar] [CrossRef]
- Bauer, A.; Jagannathan, V.; Högler, S.; Richter, B.; McEwan, N.A.; Thomas, A.; Cadieu, E.; André, C.; Hytönen, M.K.; Lohi, H.; et al. MKLN1 splicing defect in dogs with lethal acrodermatitis. PLoS Genet. 2018, 14, e1007264. [Google Scholar] [CrossRef] [Green Version]
- Murgiano, L.; Waluk, D.; Towers, R.; Wiedemar, N.; Dietrich, J.; Jagannathan, V.; Drögemüller, M.; Balmer, P.; Druet, T.; Galichet, A.; et al. An intronic MBTPS2 variant results in a splicing defect in horses with brindle coat texture. G3 Genes Genomes Genet. 2016, 6, 2963–2970. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Chen, M.; Bard, F.; Chen, S.; Zhou, H.; Woodley, D.; Polischuk, R.; Schekman, R.; Malhotra, V. TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell 2009, 136, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Peotter, J.; Kasberg, W.; Pustova, I.; Audhya, A. COPII-mediated trafficking at the ER/ERGIC interface. Traffic 2019, 20, 491–503. [Google Scholar] [CrossRef]
- Miller, E.A.; Schekman, R. COPII—A flexible vesicle formation system. Curr. Opin. Cell Biol. 2013, 25, 420–427. [Google Scholar] [CrossRef]
- Malhotra, V.; Erlmann, P. The pathway of collagen secretion. Annu. Rev. Cell Dev. Biol. 2015, 31, 109–124. [Google Scholar] [CrossRef]
- Saito, K.; Maeda, M. Not just a cargo receptor for large cargoes; an emerging role of TANGO1 as an organizer of ER exit sites. J. Biochem. 2019, 166, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.G.; Phamluong, K.; Li, L.; Sun, M.; Cao, T.C.; Liu, P.S.; Modrusan, Z.; Sandoval, W.N.; Rangell, L.; Carano, R.A.D.; et al. Global defects in collagen secretion in a Mia3/TANGO1 knockout mouse. J. Cell Biol. 2011, 193, 935–951. [Google Scholar] [CrossRef] [Green Version]
- Guillemyn, B.; Nampoothiri, S.; Syx, D.; Malfait, F.; Symoens, S. Loss of TANGO1 Leads to Absence of Bone Mineralization. JBMR Plus 2021, 5, e10451. [Google Scholar] [CrossRef]
- Cauwels, R.G.E.C.; De Coster, P.J.; Mortier, G.R.; Marks, L.A.M.; Martens, L.C. Dentinogenesis imperfecta associated with short stature, hearing loss and mental retardation: A new syndrome with autosomal recessive inheritance? J. Oral Pathol. Med. 2005, 34, 444–446. [Google Scholar] [CrossRef]
- Lekszas, C.; Foresti, O.; Raote, I.; Liedtke, D.; König, E.-M.; Nanda, I.; Vona, B.; De Coster, P.; Cauwels, R.; Malhotra, V.; et al. Biallelic TANGO1 mutations cause a novel syndromal disease due to hampered cellular collagen secretion. eLife 2020, 9, e51319. [Google Scholar] [CrossRef]
- Campbell, B.G.; Wootton, J.A.M.; MacLeod, J.N.; Minor, R.R. Sequence of normal canine COL1A1 cDNA and identification of a heterozygous α 1(I) collagen Gly208Ala mutation in a severe case of canine osteogenesis imperfecta. Arch. Biochem. Biophys. 2000, 384, 37–46. [Google Scholar] [CrossRef]
- Campbell, B.G.; Wootton, J.A.M.; Macleod, J.N.; Minor, R.R. Canine COL1A2 mutation resulting in C-terminal truncation of pro-α 2(I) and severe osteogenesis imperfecta. J. Bone Miner. Res. 2001, 16, 1147–1153. [Google Scholar] [CrossRef]
- Quist, E.M.; Doan, R.; Pool, R.R.; Porter, B.F.; Bannasch, D.L.; Dindot, S.V. Identification of a candidate mutation in the COL1A2 gene of a Chow Chow with osteogenesis imperfecta. J. Hered. 2018, 109, 308–314. [Google Scholar] [CrossRef]
- Letko, A.; Zdora, I.; Hitzler, V.; Jagannathan, V.; Beineke, A.; Möhrke, C.; Drögemüller, C. A de novo in-frame duplication in the COL1A2 gene in a Lagotto Romagnolo dog with osteogenesis imperfecta. Anim. Genet. 2019, 50, 786–787. [Google Scholar] [CrossRef]
- Drögemüller, C.; Becker, D.; Brunner, A.; Haase, B.; Kircher, P.; Seeliger, F.; Fehr, M.; Baumann, U.; Lindblad-Toh, K.; Leeb, T. A missense mutation in the SERPINH1 gene in Dachshunds with osteogenesis imperfecta. PLoS Genet. 2009, 5, e1000579. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Filtering Step | Homozygous Variants |
|---|---|
| All variants | 2,286,318 |
| Private variants | 1776 |
| With SnpEff impact high, moderate or low | 8 |
| In critical intervals | 1 |
| Phenotype | wt/wt | wt/del | del/del |
|---|---|---|---|
| DSRA cases (n = 18) | - | - | 18 |
| Non-affected control dogs (n = 17) | 8 | 9 | - |
| Dogs with unknown phenotype (n = 5) | 5 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christen, M.; Booij-Vrieling, H.; Oksa-Minalto, J.; de Vries, C.; Kehl, A.; Jagannathan, V.; Leeb, T. MIA3 Splice Defect in Cane Corso Dogs with Dental-Skeletal-Retinal Anomaly (DSRA). Genes 2021, 12, 1497. https://doi.org/10.3390/genes12101497
Christen M, Booij-Vrieling H, Oksa-Minalto J, de Vries C, Kehl A, Jagannathan V, Leeb T. MIA3 Splice Defect in Cane Corso Dogs with Dental-Skeletal-Retinal Anomaly (DSRA). Genes. 2021; 12(10):1497. https://doi.org/10.3390/genes12101497
Chicago/Turabian StyleChristen, Matthias, Henriëtte Booij-Vrieling, Jelena Oksa-Minalto, Cynthia de Vries, Alexandra Kehl, Vidhya Jagannathan, and Tosso Leeb. 2021. "MIA3 Splice Defect in Cane Corso Dogs with Dental-Skeletal-Retinal Anomaly (DSRA)" Genes 12, no. 10: 1497. https://doi.org/10.3390/genes12101497
APA StyleChristen, M., Booij-Vrieling, H., Oksa-Minalto, J., de Vries, C., Kehl, A., Jagannathan, V., & Leeb, T. (2021). MIA3 Splice Defect in Cane Corso Dogs with Dental-Skeletal-Retinal Anomaly (DSRA). Genes, 12(10), 1497. https://doi.org/10.3390/genes12101497