Identifying Risk Genes and Interpreting Pathogenesis for Parkinson’s Disease by a Multiomics Analysis



Abstract
1. Introduction
2. Materials and Methods
2.1. GWAS Analysis of Whole-Genome Data from PPMI
2.2. Meta-Analysis Based on Effect Size
2.3. Summary Data-Based Mendelian Randomization Analysis
2.4. Enrichment Test of Functional Categories
3. Results
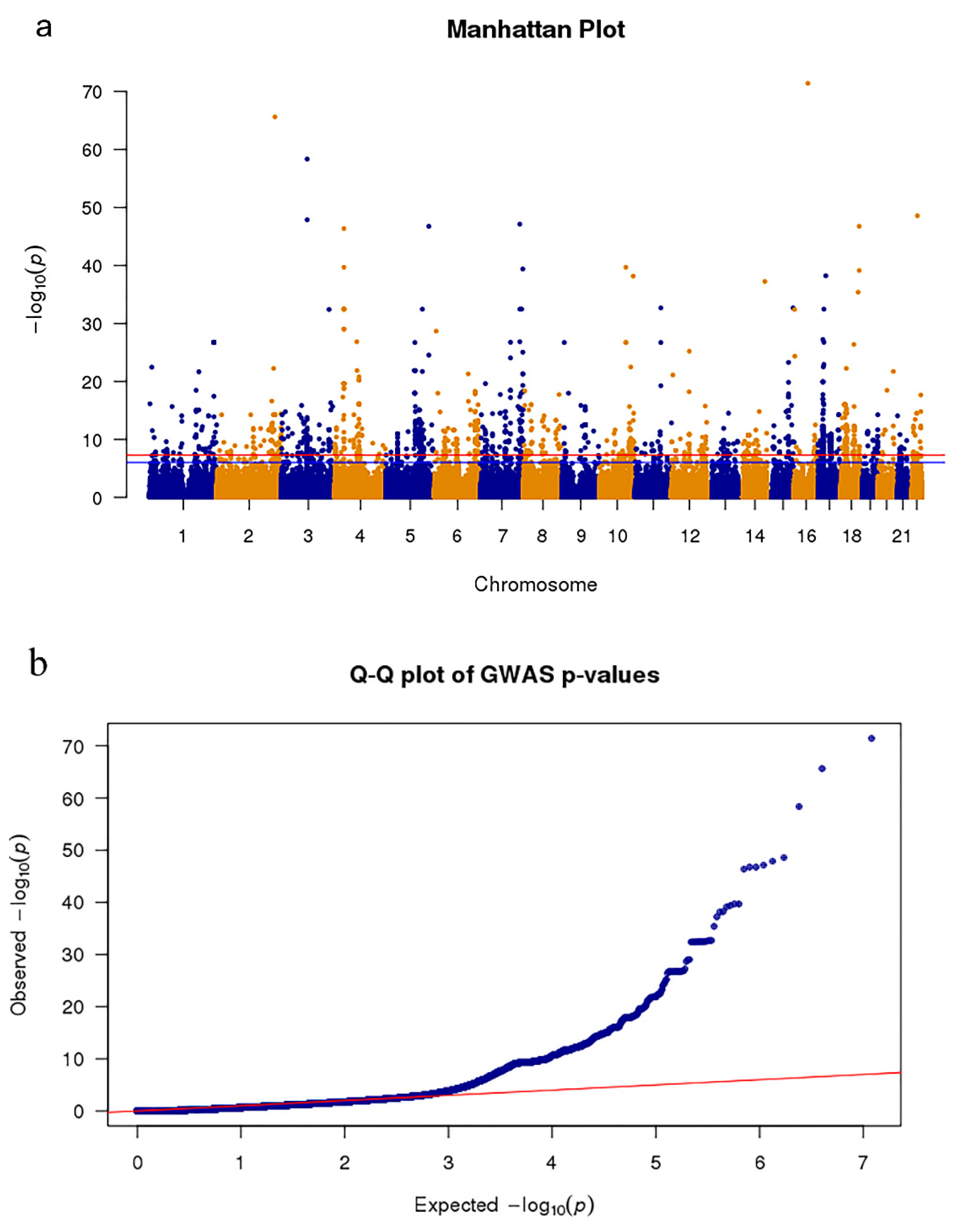
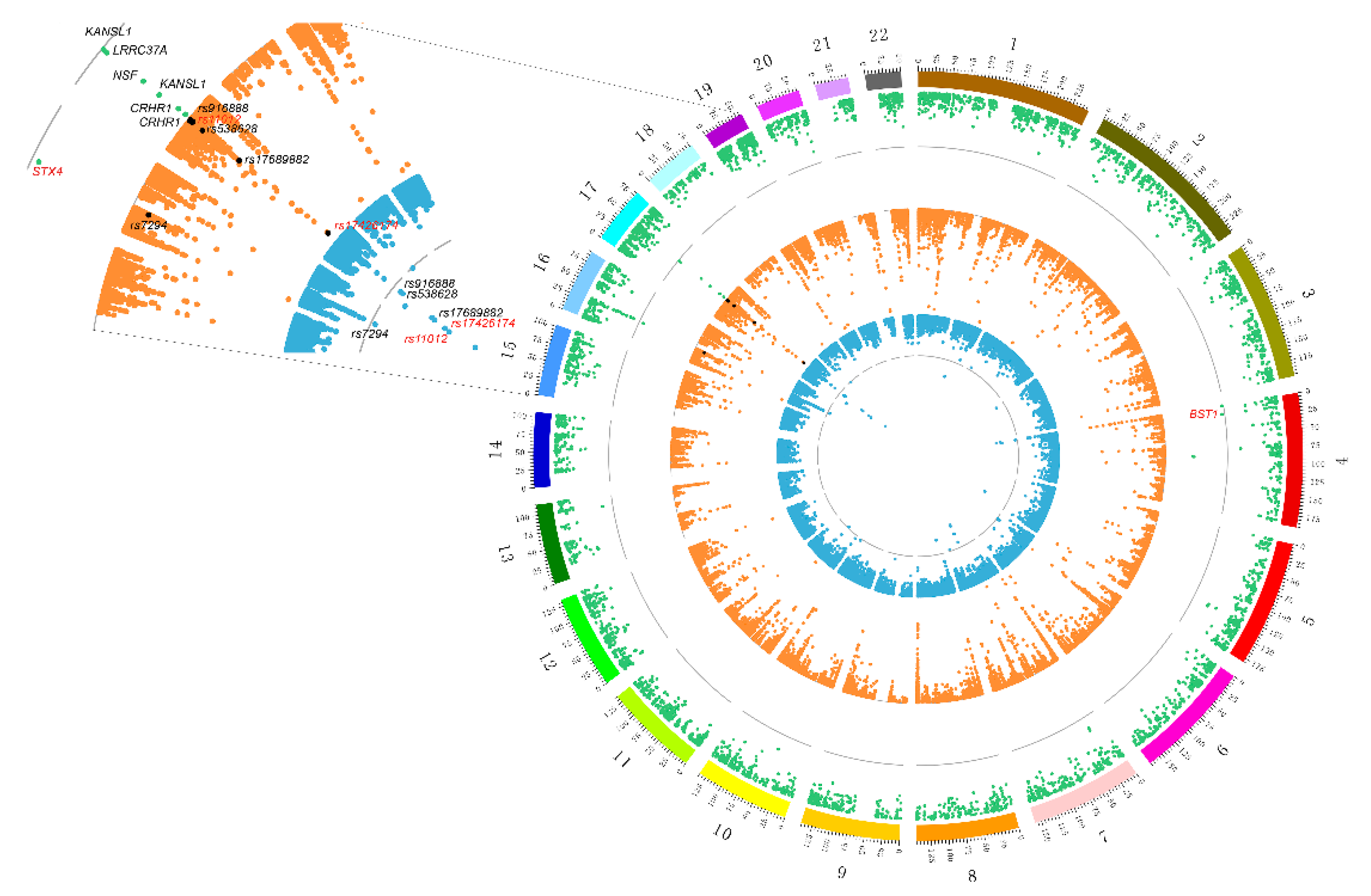
3.1. Screening SNPs of PD Based on Meta-Analysis GWAS Data
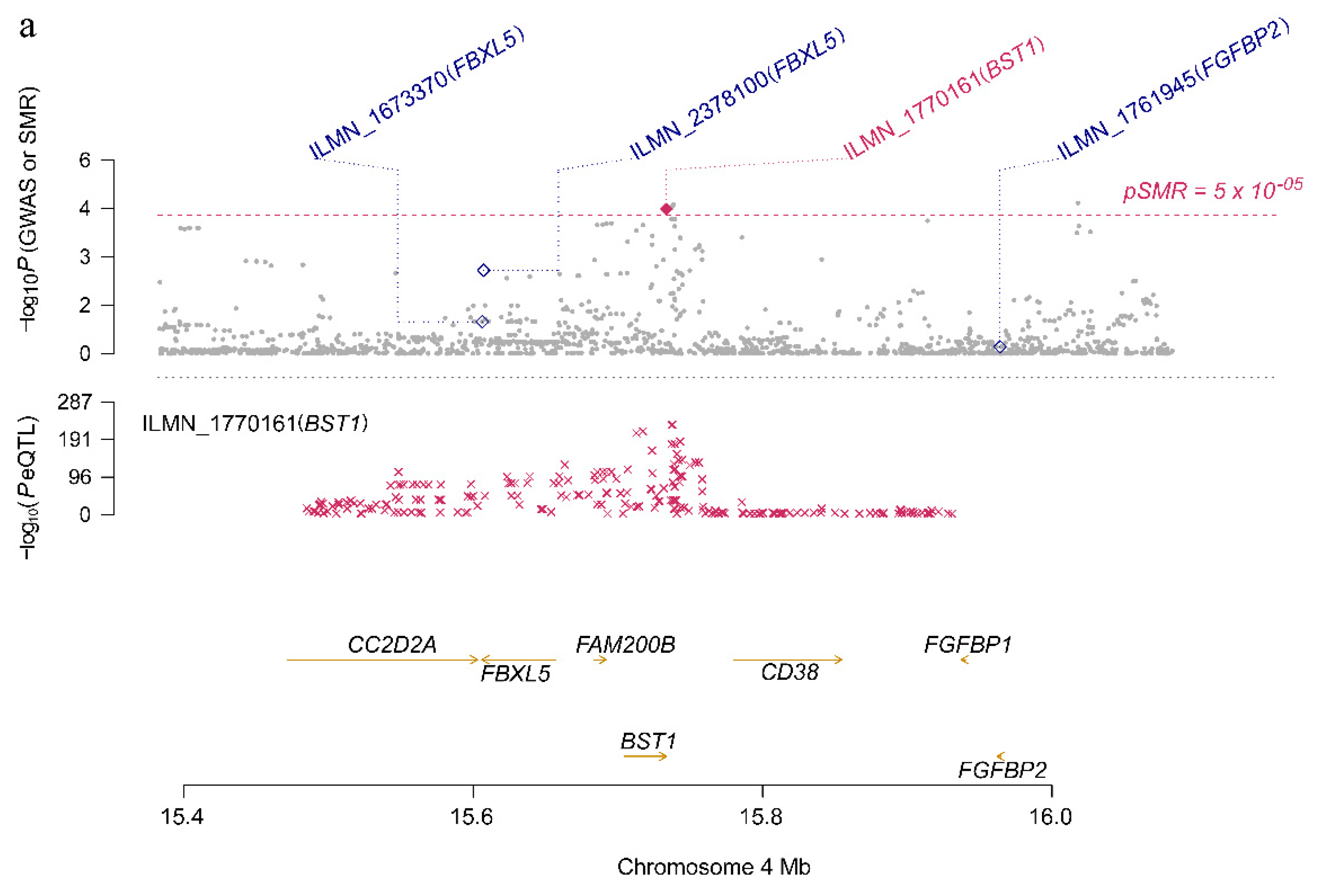
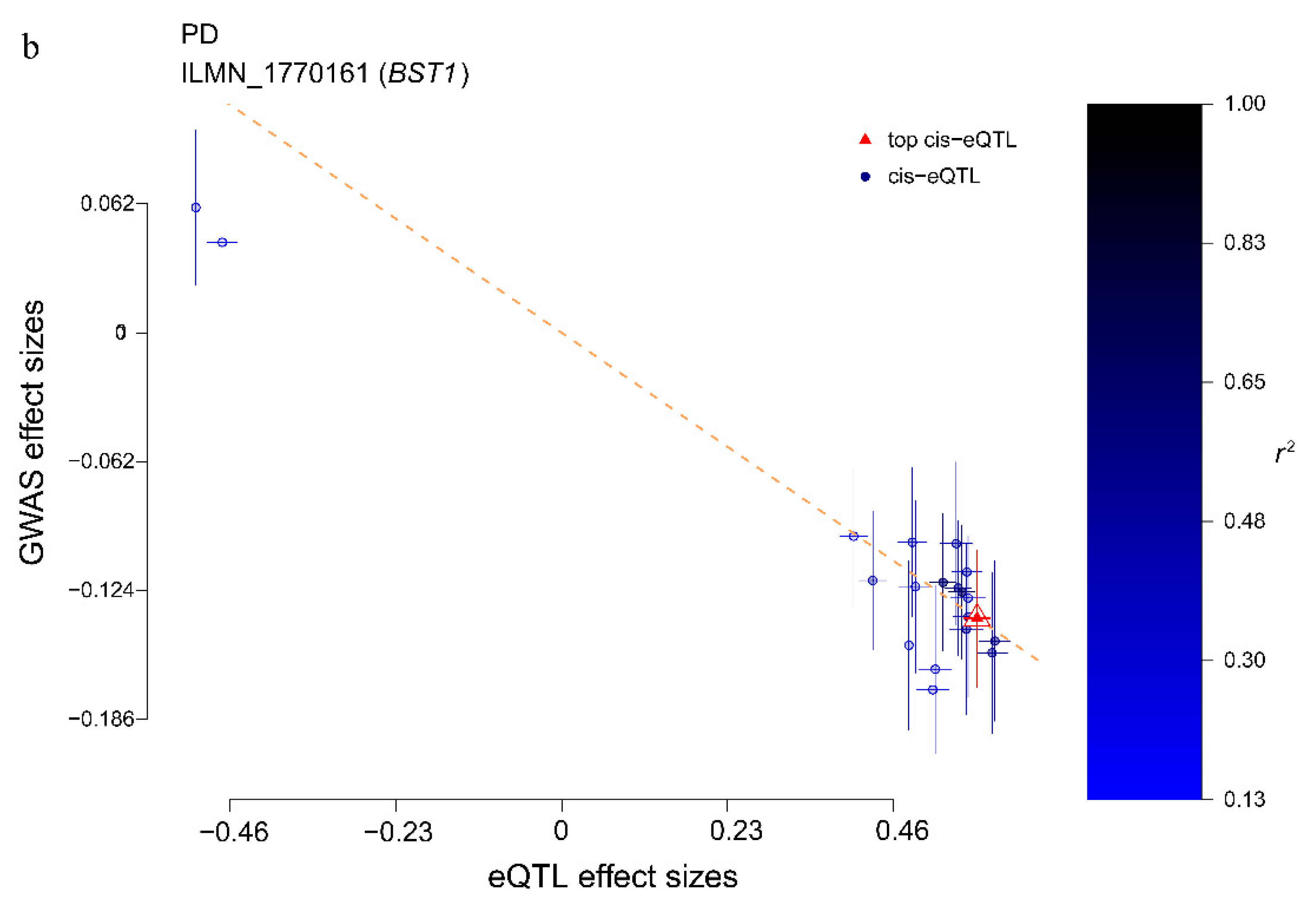
3.2. Identifying PD Risk Genes Based on GWAS and eQTL Data
3.3. Elucidating the Pathogenesis of Two Risk Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic-A Call to Action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Zheng, X.M.; Chen, J.; Pang, H.B.; Liu, S.; Gao, Q.; Wang, J.R.; Qiao, W.H.; Wang, H.; Liu, J.; Olsen, K.M.; et al. Genome-wide analyses reveal the role of noncoding variation in complex traits during rice domestication. Sci. Adv. 2019, 5, eaax3619. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.K.; Loh, P.R.; Finucane, H.K.; Ripke, S.; Yang, J.; Schizophrenia Working Group of the Psychiatric Genomics Consortium; Patterson, N.; Daly, M.J.; Price, A.L.; Neale, B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef]
- Wu, Y.; Zheng, Z.; Visscher, P.M.; Yang, J. Quantifying the mapping precision of genome-wide association studies using whole-genome sequencing data. Genome Biol. 2017, 18, 86. [Google Scholar] [CrossRef]
- Farh, K.K.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, J.; Zhang, F.; Zhu, Z.; Qi, T.; Zheng, Z.; Lloyd-Jones, L.R.; Marioni, R.E.; Martin, N.G.; Montgomery, G.W.; et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun. 2018, 9, 918. [Google Scholar] [CrossRef]
- Rueger, S.; McDaid, A.; Kutalik, Z. Evaluation and application of summary statistic imputation to discover new height-associated loci. PLoS Genet. 2018, 14, e1007371. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Jones, L.R.; Holloway, A.; McRae, A.; Yang, J.; Small, K.; Zhao, J.; Zeng, B.; Bakshi, A.; Metspalu, A.; Dermitzakis, M.; et al. The genetic architecture of gene expression in peripheral blood. Am. J. Hum. Genet. 2017, 100, 371. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Marek, K.; Chowdhury, S.; Siderowf, A.; Lasch, S.; Coffey, C.S.; Caspell-Garcia, C.; Simuni, T.; Jennings, D.; Tanner, C.M.; Trojanowski, J.Q.; et al. The Parkinson’s progression markers initiative (PPMI)—Establishing a PD biomarker cohort. Ann. Clin. Transl. Neurol. 2018, 5, 1460–1477. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Li, Y.; Abecasis, G.R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010, 26, 2190–2191. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K.; Berisa, T.; Liu, J.Z.; Segurel, L.; Tung, J.Y.; Hinds, D.A. Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 2016, 48, 709–717. [Google Scholar] [CrossRef]
- Westra, H.J.; Peters, M.J.; Esko, T.; Yaghootkar, H.; Schurmann, C.; Kettunen, J.; Christiansen, M.W.; Fairfax, B.P.; Schramm, K.; Powell, J.E.; et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef]
- Powell, J.E.; Henders, A.K.; McRae, A.F.; Caracella, A.; Smith, S.; Wright, M.J.; Whitfield, J.B.; Dermitzakis, E.T.; Martin, N.G.; Visscher, P.M.; et al. The brisbane systems genetics study: Genetical genomics meets complex trait genetics. PLoS ONE 2012, 7, e35430. [Google Scholar] [CrossRef]
- Chen, B.H.; Marioni, R.E.; Colicino, E.; Peters, M.J.; Horvath, S. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [CrossRef]
- Pankratz, N.; Beecham, G.W.; DeStefano, A.L.; Dawson, T.M.; Doheny, K.F.; Factor, S.A.; Hamza, T.H.; Hung, A.Y.; Hyman, B.T.; Ivinson, A.J.; et al. Meta-analysis of Parkinson’s disease: Identification of a novel locus, RIT2. Ann. Neurol. 2012, 71, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Pelz, J.O.; Belau, E.; Fricke, C.; Classen, J.; Weise, D. Axonal degeneration of the vagus nerve in Parkinson’s disease-a high-resolution ultrasound study. Front. Neurol. 2018, 9, 951. [Google Scholar] [CrossRef] [PubMed]
- Corti, O. Neuronal mitophagy: Lessons from a pathway linked to Parkinson’s disease. Neurotox Res. 2019, 36, 292–305. [Google Scholar] [CrossRef]
- Elsworth, J.D.; Leranth, C.; Redmond, D.E., Jr.; Roth, R.H. Loss of asymmetric spine synapses in prefrontal cortex of motor-asymptomatic, dopamine-depleted, cognitively impaired MPTP-treated monkeys. Int. J. Neuropsychopharmacol. 2013, 16, 905–912. [Google Scholar] [CrossRef][Green Version]
- Shetty, A.K.; Bates, A. Potential of GABA-ergic cell therapy for schizophrenia, neuropathic pain, and Alzheimer’s and Parkinson’s diseases. Brain Res. 2016, 1638, 74–87. [Google Scholar] [CrossRef]
- Witoelar, A.; Jansen, I.E.; Wang, Y.; Desikan, R.S.; Gibbs, J.R.; Blauwendraat, C.; Thompson, W.K.; Hernandez, D.G.; Djurovic, S.; Schork, A.J.; et al. Genome-wide pleiotropy between Parkinson disease and Autoimmune diseases. JAMA Neurol. 2017, 74, 780–792. [Google Scholar] [CrossRef]
- Ferrari, R.; Wang, Y.; Vandrovcova, J.; Guelfi, S.; Witeolar, A.; Karch, C.M.; Schork, A.J.; Fan, C.C.; Brewer, J.B.; International, F.T.D.G.C.; et al. Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J. Neurol. Neurosurg. Psychiatry 2017, 88, 152–164. [Google Scholar] [CrossRef]
- Latourelle, J.C.; Dumitriu, A.; Hadzi, T.C.; Beach, T.G.; Myers, R.H. Evaluation of Parkinson disease risk variants as expression-QTLs. PLoS ONE 2012, 7, e46199. [Google Scholar] [CrossRef]
- Foo, J.N.; Tan, L.C.; Irwan, I.D.; Au, W.L.; Low, H.Q.; Prakash, K.M.; Ahmad-Annuar, A.; Bei, J.; Chan, A.Y.; Chen, C.M.; et al. Genome-wide association study of Parkinson’s disease in East Asians. Hum. Mol. Genet. 2017, 26, 226–232. [Google Scholar] [CrossRef]
- Zhu, X.C.; Cao, L.; Tan, M.S.; Jiang, T.; Wang, H.F.; Lu, H.; Tan, C.C.; Zhang, W.; Tan, L.; Yu, J.T. Association of Parkinson’s Disease GWAS-Linked Loci with Alzheimer’s Disease in Han Chinese. Mol. Neurobiol. 2017, 54, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Lill, C.M.; Roehr, J.T.; McQueen, M.B.; Kavvoura, F.K.; Bagade, S.; Schjeide, B.M.; Schjeide, L.M.; Meissner, E.; Zauft, U.; Allen, N.C.; et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLoS Genet. 2012, 8, e1002548. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Luo, J.; Liu, L.; Fu, H.; Tang, L. The association between CD157/BST1 polymorphisms and the susceptibility of Parkinson’s disease: A meta-analysis. Neuropsychiatr. Dis. Treat. 2019, 15, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Kasai, S.; Yoshihara, T.; Lopatina, O.; Ishihara, K.; Higashida, H. Selegiline ameliorates Depression-like behavior in mice lacking the CD157/BST1 gene, a risk factor for Parkinson’s disease. Front. Behav. Neurosci. 2017, 11, 75. [Google Scholar] [CrossRef]
- Weber, H.; Richter, J.; Straube, B.; Lueken, U.; Domschke, K.; Schartner, C.; Klauke, B.; Baumann, C.; Pane-Farre, C.; Jacob, C.P.; et al. Allelic variation in CRHR1 predisposes to panic disorder: Evidence for biased fear processing. Mol. Psychiatry 2016, 21, 813–822. [Google Scholar] [CrossRef]
- Koutmani, Y.; Politis, P.K.; Elkouris, M.; Agrogiannis, G.; Kemerli, M.; Patsouris, E.; Remboutsika, E.; Karalis, K.P. Corticotropin-releasing hormone exerts direct effects on neuronal progenitor cells: Implications for neuroprotection. Mol. Psychiatry 2013, 18, 300–307. [Google Scholar] [CrossRef]
- Short, A.K.; Maras, P.M.; Pham, A.L.; Ivy, A.S.; Baram, T.Z. Blocking CRH receptors in adults mitigates age-related memory impairments provoked by early-life adversity. Neuropsychopharmacology 2020, 45, 515–523. [Google Scholar] [CrossRef]
- Ham, S.; Lee, Y.I.; Jo, M.; Kim, H.; Kang, H.; Jo, A.; Lee, G.H.; Mo, Y.J.; Park, S.C.; Lee, Y.S.; et al. Hydrocortisone-induced parkin prevents dopaminergic cell death via CREB pathway in Parkinson’s disease model. Sci. Rep. 2017, 7, 525. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrimsdottir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; Kerchner, G.A.; Ayalon, G.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- Schork, A.J.; Thompson, W.K.; Pham, P.; Torkamani, A.; Roddey, J.C.; Sullivan, P.F.; Kelsoe, J.R.; O’Donovan, M.C.; Furberg, H.; The Tobacco and Genetics Consortium; et al. All SNPs are not created equal: Genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS Genet. 2013, 9, e1003449. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H.; Li, Y.S.; Shi, M.M.; Yang, J.; Liu, Y.T.; Mao, C.Y.; Fan, Y.; Hu, X.C.; Shi, C.H.; Xu, Y.M. SNCA but not DNM3 and GAK modifies age at onset of LRRK2-related Parkinson’s disease in Chinese population. J. Neurol. 2019, 266, 1796–1800. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Ikuno, M.; Hondo, M.; Parajuli, L.K.; Taguchi, K.; Ueda, J.; Sawamura, M.; Okuda, S.; Nakanishi, E.; Hara, J.; et al. alpha-Synuclein BAC transgenic mice exhibit RBD-like behaviour and hyposmia: A prodromal Parkinson’s disease model. Brain 2020, 143, 249–265. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for ‘Omics’ research on drugs. Nucleic Acids Res. 2010, 39, D1035–D1041. [Google Scholar] [CrossRef]
- McKenzie, M.; Henders, A.K.; Caracella, A.; Wray, N.R.; Powell, J.E. Overlap of expression quantitative trait loci (eQTL) in human brain and blood. BMC Med. Genom. 2014, 7, 31. [Google Scholar] [CrossRef]
- Qi, T.; Wu, Y.; Zeng, J.; Zhang, F.; Xue, A.; Jiang, L.; Zhu, Z.; Kemper, K.; Yengo, L.; Zheng, Z.; et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat. Commun. 2018, 9, 2282. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
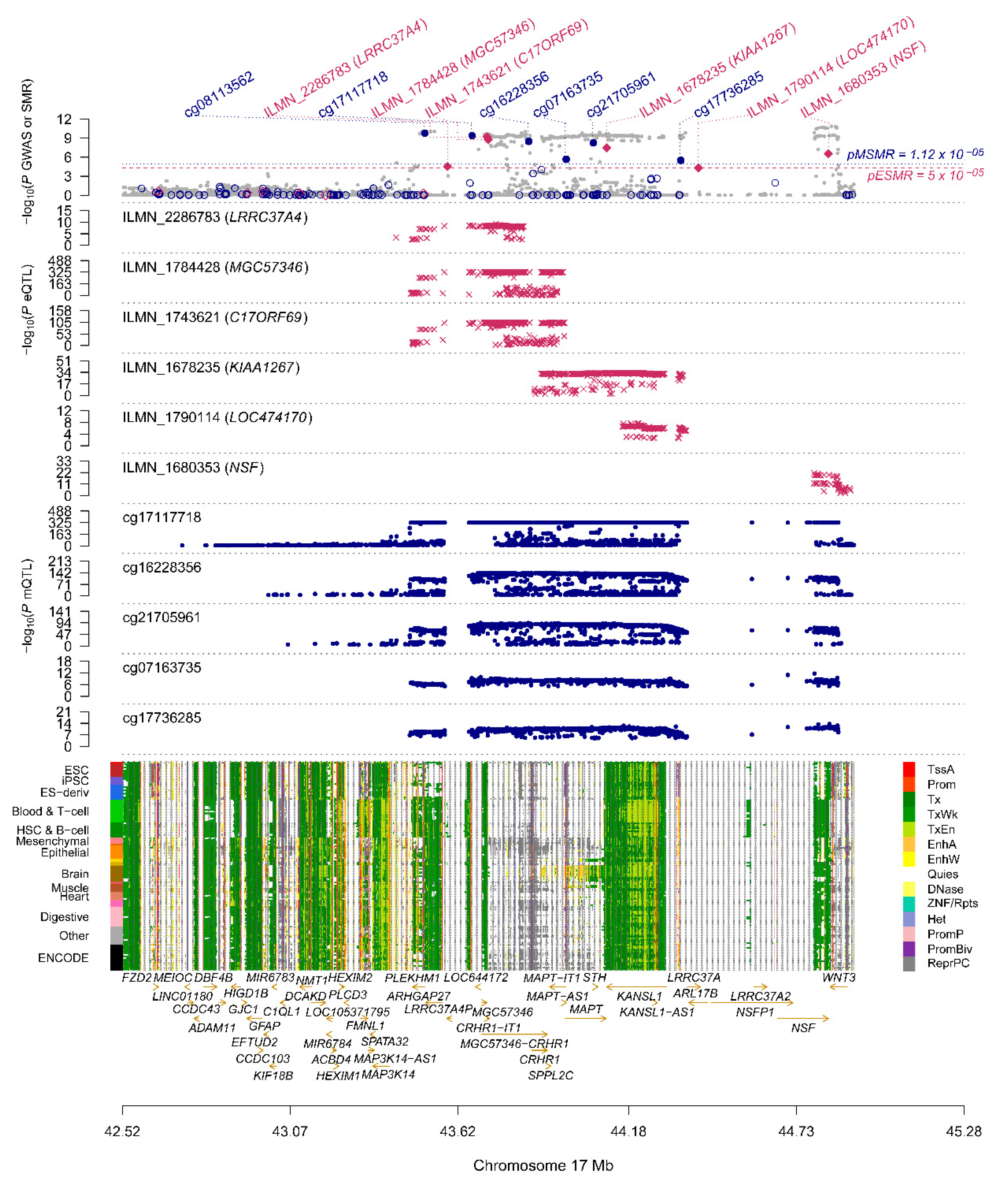
| ProbeID | Chr | Gene | topSNP | A1 | PGWAS | FDR PSMR | PHEIDI |
|---|---|---|---|---|---|---|---|
| ILMN_1784428 | 17 | MGC57346 (CRHR1) | rs4074462 | T | 4.2 × 10−10 | 3.91 × 10−6 | 0.605 |
| ILMN_1743621 | 17 | C17ORF69 (CRHR1) | rs17426195 | A | 3.53 × 10−10 | 4.91 × 10−6 | 0.507 |
| ILMN_1678235 | 17 | KIAA1267 (KANSL1) | rs4630591 | T | 5.59 × 10−10 | 6.43 × 10−5 | 0.989 |
| ILMN_1680353 | 17 | NSF | rs183211 | A | 1.24 × 10−9 | 3 × 10−4 | 0.036 |
| ILMN_1680313 | 16 | STX4 | rs8056842 | C | 1.39 × 10−5 | 2.14 × 10−4 | 0.732 |
| ILMN_2286783 | 17 | LRRC37A4 (LRRC37A) | rs1635298 | T | 1.56 × 10−8 | 2.14 × 10−4 | 0.225 |
| ILMN_1770161 | 4 | BST1 | rs4698412 | G | 2.83 × 10−5 | 2.14 × 10−4 | 0.738 |
| ILMN_1790114 | 17 | LOC474170 (KANSL1) | rs9915547 | C | 2.85 × 10−9 | 2.48 × 10−4 | 0.551 |
| Methylation (M)—Trait (T) | Methylation—Expression (E) | Expression—Trait | ||||||
|---|---|---|---|---|---|---|---|---|
| Mprobel_Region | Mprobe | M_SNP | MT_bSMR | Eprobe | E_SNP | ME_bSMR | Gene | ET_bSMR |
| Prom | cg17117718 | rs17426174 | −0.17 | ILMN_1743621 | rs17426174 | 0.33 | C17ORF69 (CRHR1) | −0.51 |
| ReprPC | cg16228356 | rs17689882 | 0.27 | rs17689882 | −0.54 | |||
| TxWk | cg21705961 | rs17689882 | 0.34 | rs17689882 | −0.67 | |||
| TssA | cg07163735 | rs538628 | −0.97 | rs17689882 | 2.25 | |||
| Quies | cg17736285 | rs916888 | 0.88 | rs1876831 | −1.88 | |||
| Prom | cg08113562 | rs11012 | −0.22 | ILMN_1678235 | rs16940665 | −0.26 | KIAA1267 (KANSL1) | 0.88 |
| Prom | cg17117718 | rs17426174 | −0.17 | rs16940665 | −0.18 | |||
| ReprPC | cg16228356 | rs17689882 | 0.27 | rs17689882 | 0.31 | |||
| TxWk | cg21705961 | rs17689882 | 0.34 | rs17689882 | 0.38 | |||
| Quies | cg17736285 | rs916888 | 0.88 | rs17575507 | 1.07 | |||
| Prom | cg08113562 | rs11012 | −0.22 | ILMN_2286783 | rs11012 | −0.10 | LRRC37A4 (LRRC37A) | |
| Prom | cg17117718 | rs17426174 | −0.17 | rs17426174 | −0.09 | 1.58 | ||
| ReprPC | cg16228356 | rs17689882 | 0.27 | rs17426174 | 0.15 | |||
| TxWk | cg21705961 | rs17689882 | 0.34 | rs17426174 | 0.19 | |||
| TssA | cg07163735 | rs538628 | −0.97 | rs17426174 | −0.63 | |||
| Quies | cg17736285 | rs916888 | 0.88 | rs17426174 | 0.54 | |||
| Prom | cg17117718 | rs17426174 | −0.17 | ILMN_1784428 | rs17426174 | 0.57 | MGC57346 (CRHR1) | −0.3 |
| ReprPC | cg16228356 | rs17689882 | 0.27 | rs17689882 | −0.93 | |||
| TxWk | cg21705961 | rs17689882 | 0.34 | rs17689882 | −1.16 | |||
| TssA | cg07163735 | rs538628 | −0.97 | rs17689882 | 3.91 | |||
| Quies | cg17736285 | rs916888 | 0.88 | rs1876831 | −3.27 | |||
| Prom | cg08113562 | rs11012 | −0.22 | ILMN_1680353 | rs199448 | 0.17 | NSF | −1.09 |
| Prom | cg17117718 | rs17426174 | −0.17 | rs199448 | 0.12 | |||
| ReprPC | cg16228356 | rs17689882 | 0.27 | rs199448 | −0.2 | |||
| TxWk | cg21705961 | rs17689882 | 0.34 | rs538628 | −0.25 | |||
| TssA | cg07163735 | rs538628 | −0.97 | rs538628 | 0.62 | |||
| Quies | cg17736285 | rs916888 | 0.88 | rs916888 | −0.62 | |||
| Tx | cg01067137 | rs7294 | 0.29 | ILMN_1680313 | rs7294 | −0.57 | STX4 | −0.45 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, W.-W.; Zhu, Q.; Zhang, H.-Y. Identifying Risk Genes and Interpreting Pathogenesis for Parkinson’s Disease by a Multiomics Analysis. Genes 2020, 11, 1100. https://doi.org/10.3390/genes11091100
Cheng W-W, Zhu Q, Zhang H-Y. Identifying Risk Genes and Interpreting Pathogenesis for Parkinson’s Disease by a Multiomics Analysis. Genes. 2020; 11(9):1100. https://doi.org/10.3390/genes11091100
Chicago/Turabian StyleCheng, Wen-Wen, Qiang Zhu, and Hong-Yu Zhang. 2020. "Identifying Risk Genes and Interpreting Pathogenesis for Parkinson’s Disease by a Multiomics Analysis" Genes 11, no. 9: 1100. https://doi.org/10.3390/genes11091100
APA StyleCheng, W.-W., Zhu, Q., & Zhang, H.-Y. (2020). Identifying Risk Genes and Interpreting Pathogenesis for Parkinson’s Disease by a Multiomics Analysis. Genes, 11(9), 1100. https://doi.org/10.3390/genes11091100