Splicing Factor Transcript Abundance in Saliva as a Diagnostic Tool for Breast Cancer

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
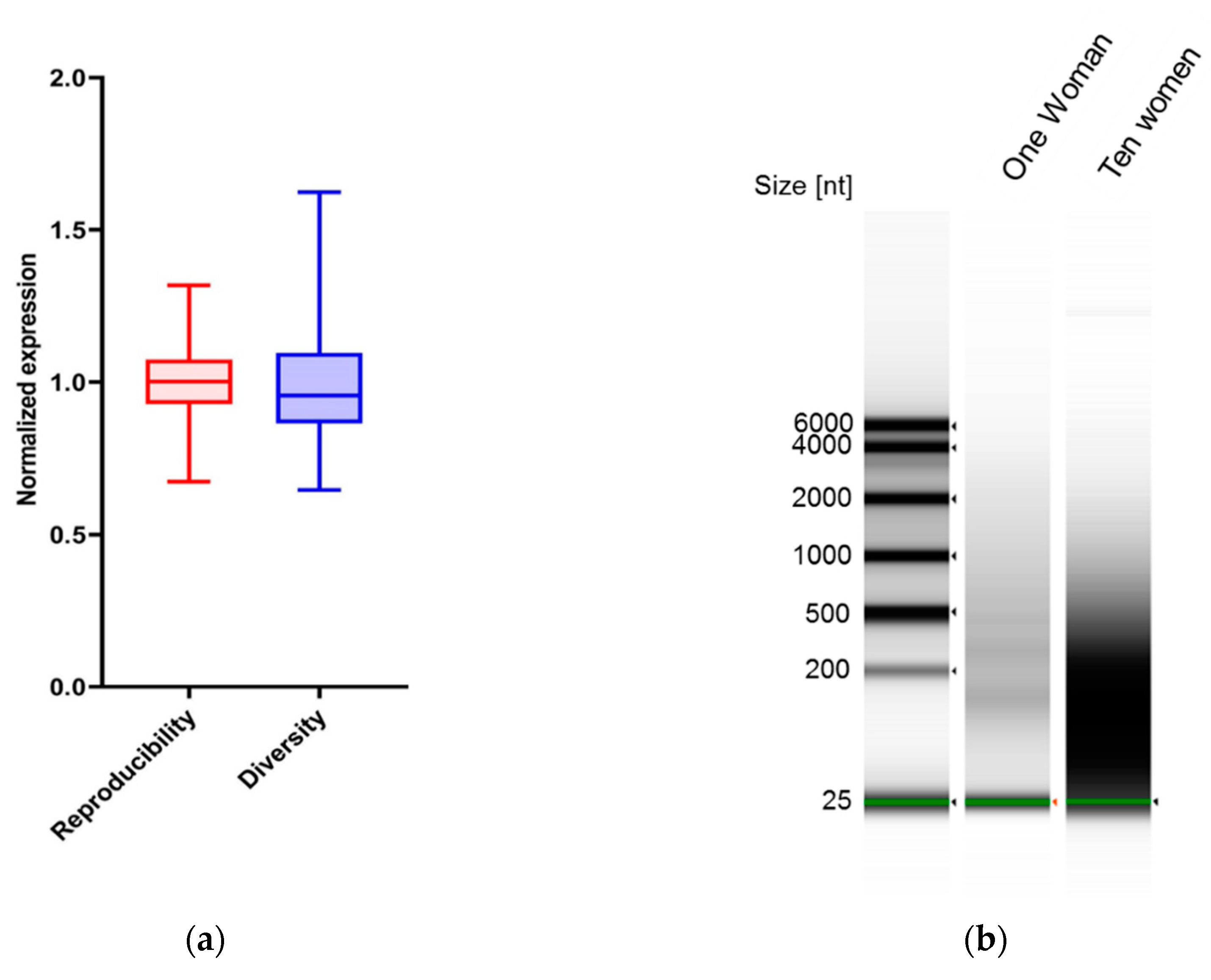
2.2. RNA Isolation and Real-Time PCR
2.3. RNA-Seq
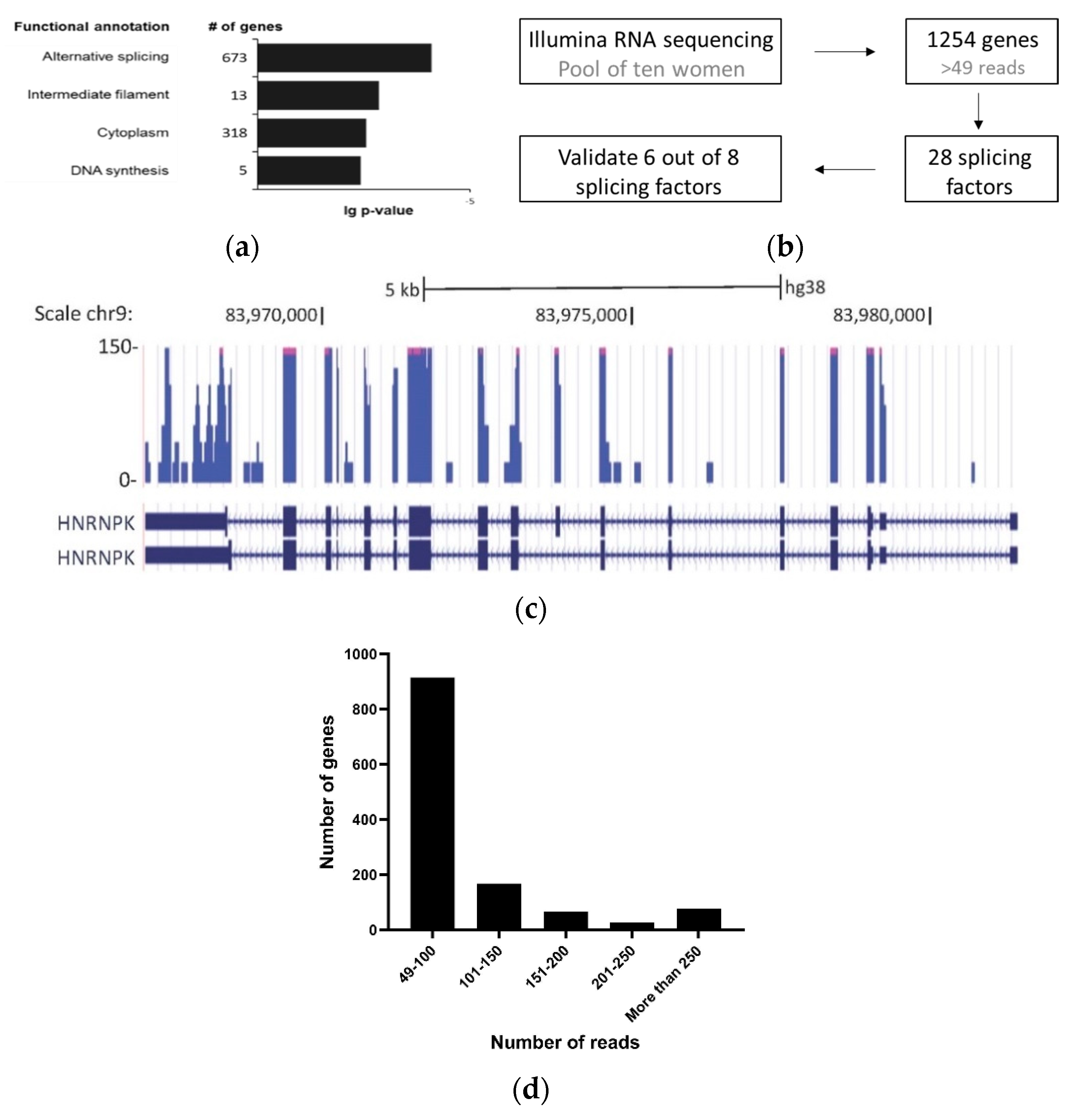
2.4. RNA-Seq Analysis
2.5. Data Availability
3. Results
3.1. Detection of Splicing Factors mRNA in Cell-Free Saliva
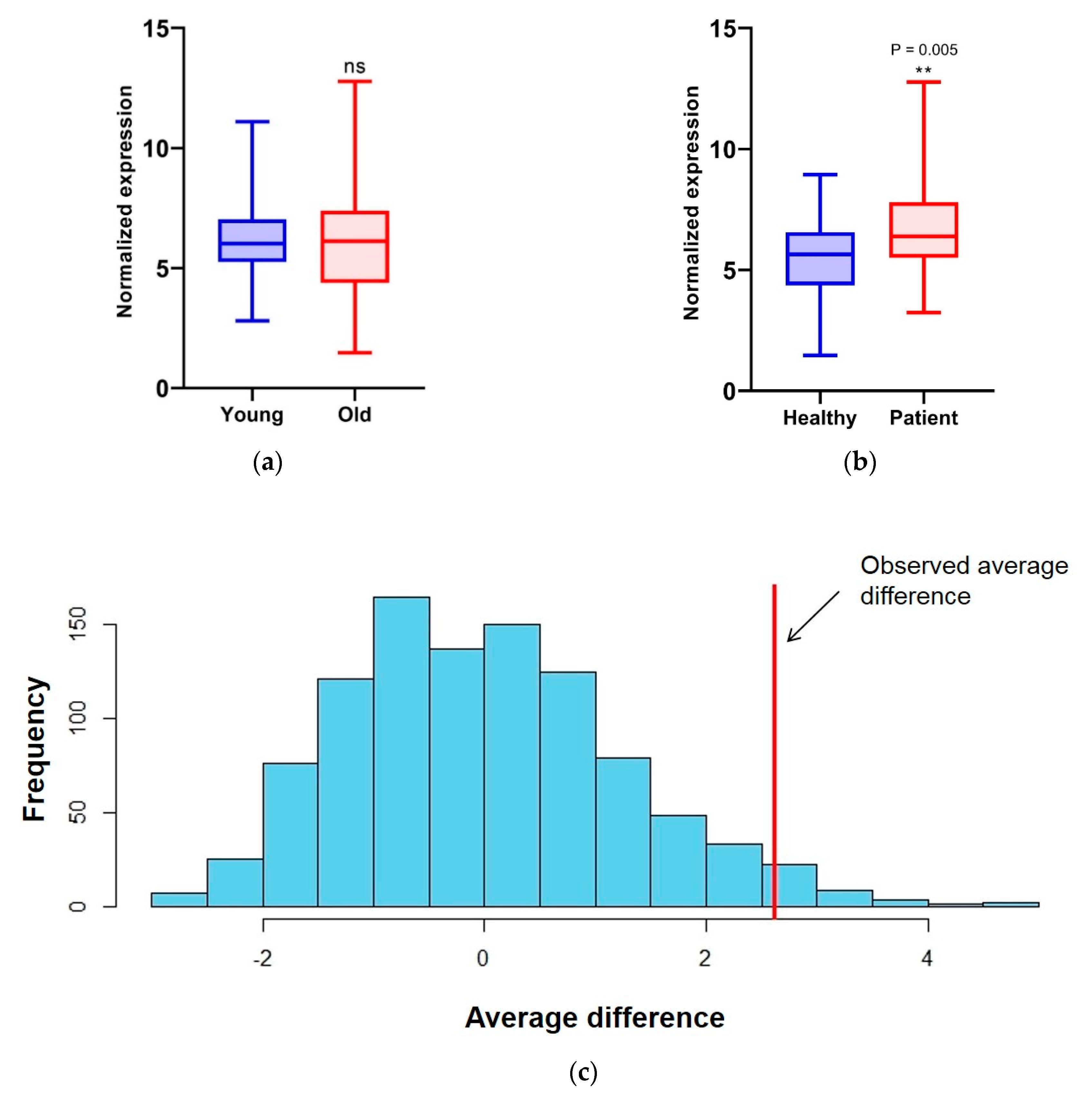
3.2. Markers in Saliva Are Consistent in the Same Woman on Different Days
3.3. Transcript Level of Splicing Factors Can Be Used as Markers for Breast Cancer in Cell-Free Saliva
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Duggan, M.A.; Anderson, W.F.; Altekruse, S.; Penberthy, L.; Sherman, M.E. The Surveillance, Epidemiology, and End Results (SEER) Program and Pathology: Toward Strengthening the Critical Relationship. Am. J. Surg. Pathol. 2016, 40, e94–e102. [Google Scholar] [CrossRef]
- Zaidi, Z.; Dib, H.A. Abstract 4191: The worldwide female breast cancer incidence and survival, 2018. Cancer Res. 2019, 79, 4191. [Google Scholar] [CrossRef]
- Breast Cancer in Women in Israel, Update of Morbidity and Mortality Data; The Israel National Cancer Registry, Israel Ministry of Health: Tel HaShomer, Ramat Gan, Israel, 2019.
- Anders, C.K.; Johnson, R.; Litton, J.; Phillips, M.; Bleyer, A. Breast cancer before age 40 years. Semin. Oncol. 2009, 36, 237–249. [Google Scholar] [CrossRef]
- World Health Organization. WHO Position Paper on Mammography Screening; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Porto-Mascarenhas, E.C.; Assad, D.X.; Chardin, H.; Gozal, D.; de Luca, C.G.; Acevedo, A.C.; Guerra, E.N. Salivary biomarkers in the diagnosis of breast cancer: A review. Crit. Rev. Oncol. Hematol. 2017, 110, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Streckfus, C.; Bigler, L.; Dellinger, T.; Dai, X.; Kingman, A.; Thigpen, J.T. The Presence of Soluble c-erbB-2 in Saliva and Serum among Women with Breast Carcinoma: A Preliminary Study. Clin. Cancer Res. 2000, 6, 2363–2370. [Google Scholar]
- Streckfus, C.; Bigler, L.; Tucci, M.; Thigpen, J.T. A Preliminary Study of CA15-3, c-erbB-2, Epidermal Growth Factor Receptor, Cathepsin-D, and p53 in Saliva among Women with Breast Carcinoma. Cancer Investig. 2000, 18, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Bigler, L.R.; Streckfus, C.F.; Copeland, L.; Burns, R.; Dai, X.; Kuhn, M.; Martin, P.; Bigler, S.A. The potential use of saliva to detect recurrence of disease in women with breast carcinoma. J. Oral Pathol. Med. 2002, 31, 421–431. [Google Scholar] [CrossRef]
- Zhang, L.; Xiao, H.; Karlan, S.; Zhou, H.; Gross, J.; Elashoff, D.; Akin, D.; Yan, X.; Chia, D.; Karlan, B.; et al. Discovery and preclinical validation of salivary transcriptomic and proteomic biomarkers for the non-invasive detection of breast cancer. PLoS ONE 2010, 5, e15573. [Google Scholar] [CrossRef]
- Sugimoto, M.; Wong, D.T.; Hirayama, A.; Soga, T.; Tomita, M. Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics 2010, 6, 78–95. [Google Scholar] [CrossRef]
- Cheng, F.; Wang, Z.; Huang, Y.; Duan, Y.; Wang, X. Investigation of salivary free amino acid profile for early diagnosis of breast cancer with ultra performance liquid chromatography-mass spectrometry. Clin. Chim. Acta 2015, 447, 23–31. [Google Scholar] [CrossRef]
- Christodoulides, N.; Mohanty, S.; Miller, C.S.; Langub, M.C.; Floriano, P.N.; Dharshan, P.; Ali, M.F.; Bernard, B.; Romanovicz, D.; Anslyn, E.; et al. Application of microchip assay system for the measurement of C-reactive protein in human saliva. Lab Chip 2005, 5, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Jia, L.; Zheng, Y.; Li, W. Salivary Exosomes: Emerging Roles in Systemic Disease. Int. J. Biol. Sci. 2018, 14, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.D.; Wu, Y.; Shen, H.Y.; Lv, M.M.; Chen, W.X.; Zhang, X.H.; Zhong, S.L.; Tang, J.H.; Zhao, J.H. Exosomes in development, metastasis and drug resistance of breast cancer. Cancer Sci. 2015, 106, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wei, F.; Schafer, C.; Wong, D.T. Detection of tumor cell-specific mRNA and protein in exosome-like microvesicles from blood and saliva. PLoS ONE 2014, 9, e110641. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Giulietti, M.; Piva, F.; D’Antonio, M.; D’Onorio, D.M.P.; Paoletti, D.; Castrignano, T.; D’Erchia, A.M.; Picardi, E.; Zambelli, F.; Principato, G.; et al. SpliceAid-F: A database of human splicing factors and their RNA-binding sites. Nucleic Acids Res. 2013, 41, D125–D131. [Google Scholar] [CrossRef]
- Shilo, A.; Siegfried, Z.; Karni, R. The role of splicing factors in deregulation of alternative splicing during oncogenesis and tumor progression. Mol. Cell. Oncol. 2015, 2, e970955. [Google Scholar] [CrossRef] [PubMed]
- Grosso, A.R.; Martins, S.; Carmo-Fonseca, M. The emerging role of splicing factors in cancer. EMBO Rep. 2008, 9, 1087–1093. [Google Scholar] [CrossRef]
- Park, S.; Brugiolo, M.; Akerman, M.; Das, S.; Urbanski, L.; Geier, A.; Kesarwani, A.K.; Fan, M.; Leclair, N.; Lin, K.T.; et al. Differential Functions of Splicing Factors in Mammary Transformation and Breast Cancer Metastasis. Cell Rep. 2019, 29, 2672–2688.e7. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Krainer, A.R.; Abdel-Wahab, O. SnapShot: Splicing Alterations in Cancer. Cell 2020, 180, 208.e1. [Google Scholar] [CrossRef] [PubMed]
- Grzybowska, E.A. Human intronless genes: Functional groups, associated diseases, evolution, and mRNA processing in absence of splicing. Biochem. Biophys. Res. Commun. 2012, 424, 1–6. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Luhrmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef]
- Graveley, B.R. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001, 17, 100–107. [Google Scholar] [CrossRef]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef]
- Salton, M.; Kasprzak, W.K.; Voss, T.; Shapiro, B.A.; Poulikakos, P.I.; Misteli, T. Inhibition of vemurafenib-resistant melanoma by interference with pre-mRNA splicing. Nat. Commun. 2015, 6, 7103. [Google Scholar] [CrossRef]
- Venables, J.P.; Klinck, R.; Bramard, A.; Inkel, L.; Dufresne-Martin, G.; Koh, C.; Gervais-Bird, J.; Lapointe, E.; Froehlich, U.; Durand, M.; et al. Identification of alternative splicing markers for breast cancer. Cancer Res. 2008, 68, 9525–9531. [Google Scholar] [CrossRef]
- Pelisch, F.; Khauv, D.; Risso, G.; Stallings-Mann, M.; Blaustein, M.; Quadrana, L.; Radisky, D.C.; Srebrow, A. Involvement of hnRNP A1 in the matrix metalloprotease-3-dependent regulation of Rac1 pre-mRNA splicing. J. Cell. Biochem. 2012, 113, 2319–2329. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Allred, D.C.; Avis, I.; Martinez, A.; Vos, M.D.; Smith, L.; Treston, A.M.; Mulshine, J.L. Differential expression of the early lung cancer detection marker, heterogeneous nuclear ribonucleoprotein-A2/B1 (hnRNP-A2/B1) in normal breast and neoplastic breast cancer. Breast Cancer Res. Treat. 2001, 66, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sun, Z.; Deng, J.; Hu, B.; Yan, W.; Wei, H.; Jiang, J. Splicing factor hnRNPA2B1 contributes to tumorigenic potential of breast cancer cells through STAT3 and ERK1/2 signaling pathway. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2017, 39, 1010428317694318. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Vadlamudi, R.; Nguyen, D.; Wang, R.A.; Costa, L.; Bagheri-Yarmand, R.; Mendelsohn, J.; Kumar, R. Growth factors regulate heterogeneous nuclear ribonucleoprotein K expression and function. J. Biol. Chem. 2001, 276, 9699–9704. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.Y.; Fu, S.L.; Tseng, L.M.; Chiu, J.H.; Lin, C.H. hnRNPK S379 phosphorylation participates in migration regulation of triple negative MDA-MB-231 cells. Sci. Rep. 2019, 9, 7611. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Fan, Y.; Yu, X.; Mao, X.; Jin, F. PTBP1 promotes the growth of breast cancer cells through the PTEN/Akt pathway and autophagy. J. Cell. Physiol. 2018, 233, 8930–8939. [Google Scholar] [CrossRef]
- He, X.; Arslan, A.D.; Ho, T.T.; Yuan, C.; Stampfer, M.R.; Beck, W.T. Involvement of polypyrimidine tract-binding protein (PTBP1) in maintaining breast cancer cell growth and malignant properties. Oncogenesis 2014, 3, e84. [Google Scholar] [CrossRef]
- Das, S.; Akerman, M.; Park, S.; Brugioli, M.; Geier, A.; Kesarwani, A.K.; Fan, M.; Leclair, N.; Urbanski, L.; Lin, K.-T.; et al. Differential Functions of Splicing Factors in Breast-Cancer Initiation and Metastasis. bioRxiv 2019, 10, 634154. [Google Scholar] [CrossRef]
- Lal, S.; Allan, A.; Markovic, D.; Walker, R.; Macartney, J.; Europe-Finner, N.; Tyson-Capper, A.; Grammatopoulos, D.K. Estrogen alters the splicing of type 1 corticotropin-releasing hormone receptor in breast cancer cells. Sci. Signal. 2013, 6, ra53. [Google Scholar] [CrossRef]
- Luqmani, Y.A.; Mortimer, C.; Yiangou, C.; Johnston, C.L.; Bansal, G.S.; Sinnett, D.; Law, M.; Coombes, R.C. Expression of 2 variant forms of fibroblast growth factor receptor 1 in human breast. Int. J. Cancer 1995, 64, 274–279. [Google Scholar] [CrossRef]
- Siam, A.; Baker, M.; Amit, L.; Regev, G.; Rabner, A.; Najar, R.A.; Bentata, M.; Dahan, S.; Cohen, K.; Araten, S.; et al. Regulation of alternative splicing by p300-mediated acetylation of splicing factors. RNA 2019, 25, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Urbanski, L.M.; Leclair, N.; Anczukow, O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev. RNA 2018, 9, e1476. [Google Scholar] [CrossRef]
- Eswaran, J.; Horvath, A.; Godbole, S.; Reddy, S.D.; Mudvari, P.; Ohshiro, K.; Cyanam, D.; Nair, S.; Fuqua, S.A.; Polyak, K.; et al. RNA sequencing of cancer reveals novel splicing alterations. Sci. Rep. 2013, 3, 1689. [Google Scholar] [CrossRef] [PubMed]
- Anczukow, O.; Rosenberg, A.Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.K.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Anczukow, O.; Akerman, M.; Clery, A.; Wu, J.; Shen, C.; Shirole, N.H.; Raimer, A.; Sun, S.; Jensen, M.A.; Hua, Y.; et al. SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol. Cell 2015, 60, 105–117. [Google Scholar] [CrossRef]
- Buoso, E.; Ronfani, M.; Galasso, M.; Ventura, D.; Corsini, E.; Racchi, M. Cortisol-induced SRSF3 expression promotes GR splicing, RACK1 expression and breast cancer cells migration. Pharmacol. Res. 2019, 143, 17–26. [Google Scholar] [CrossRef]
- Huang, C.S.; Shen, C.Y.; Wang, H.W.; Wu, P.E.; Cheng, C.W. Increased expression of SRp40 affecting CD44 splicing is associated with the clinical outcome of lymph node metastasis in human breast cancer. Clin. Chim. Acta 2007, 384, 69–74. [Google Scholar] [CrossRef]
- Watermann, D.O.; Tang, Y.; Zur Hausen, A.; Jager, M.; Stamm, S.; Stickeler, E. Splicing factor Tra2-beta1 is specifically induced in breast cancer and regulates alternative splicing of the CD44 gene. Cancer Res. 2006, 66, 4774–4780. [Google Scholar] [CrossRef]
- Moran-Jones, K.; Grindlay, J.; Jones, M.; Smith, R.; Norman, J.C. hnRNP A2 regulates alternative mRNA splicing of TP53INP2 to control invasive cell migration. Cancer Res. 2009, 69, 9219–9227. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, X.D.; Lee, J.H.; Huang, H.; Tan, H.; Ahn, J.; Reinke, L.M.; Peter, M.E.; Feng, Y.; Gius, D.; et al. Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev. 2014, 28, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, L.; Caracciolo, V.; Giordano, A. NSP 5a3a’s link to nuclear-cyto proteins B23 and hnRNP-L between normal and aberrant breast cell lines. Cell Cycle 2010, 9, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.M.; Cheng, A.W.; Flytzanis, N.C.; Balsamo, M.; Condeelis, J.S.; Oktay, M.H.; Burge, C.B.; Gertler, F.B. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011, 7, e1002218. [Google Scholar] [CrossRef] [PubMed]
- Rintala-Maki, N.D.; Goard, C.A.; Langdon, C.E.; Wall, V.E.; Traulsen, K.E.; Morin, C.D.; Bonin, M.; Sutherland, L.C. Expression of RBM5-related factors in primary breast tissue. J. Cell. Biochem. 2007, 100, 1440–1458. [Google Scholar] [CrossRef]
- Song, L.; Wang, L.; Li, Y.; Xiong, H.; Wu, J.; Li, J.; Li, M. Sam68 up-regulation correlates with, and its down-regulation inhibits, proliferation and tumourigenicity of breast cancer cells. J. Pathol. 2010, 222, 227–237. [Google Scholar] [CrossRef]
- Venables, J.P.; Klinck, R.; Koh, C.; Gervais-Bird, J.; Bramard, A.; Inkel, L.; Durand, M.; Couture, S.; Froehlich, U.; Lapointe, E.; et al. Cancer-associated regulation of alternative splicing. Nat. Struct. Mol. Biol. 2009, 16, 670–676. [Google Scholar] [CrossRef]
- Oeffinger, K.C.; Fontham, E.T.H.; Etzioni, R.; Herzig, A.; Michaelson, J.S.; Shih, Y.-C.T.; Walter, L.C.; Church, T.R.; Flowers, C.R.; LaMonte, S.J.; et al. Breast Cancer Screening for Women at Average Risk: 2015 Guideline Update From the American Cancer Society. JAMA 2015, 314, 1599–1614. [Google Scholar] [CrossRef]
- Yankaskas, B.C.; Haneuse, S.; Kapp, J.M.; Kerlikowske, K.; Geller, B.; Buist, D.S.; Breast Cancer Surveillance Consortium. Performance of first mammography examination in women younger than 40 years. J. Natl. Cancer Inst. 2010, 102, 692–701. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| # | Age (Years) at: | IDC 1 | ILC 2 | HER2 | Site of Metastasis | Treatment | ||
|---|---|---|---|---|---|---|---|---|
| Collection | Diagnosis | Death | ||||||
| 1 | 66 | 43 | 67 | √ | √ | + | Liver, lung, bone | Chemotherapy (carboplatin/gemzar) |
| 2 | 57 | 56 | 58 | √ | × | − | Liver, bone | Hormonal and Palbocyclib |
| 3 | 36 | 27 | 37 | √ | × | − | Bone, lung | Chemotherapy (cyclophosphamide/methotrexate/5FU 3) |
| 4 | 55 | 54 | √ | × | + | Local disease | Chemotherapy (adriamycin/cyclophosphamide (neoadjuvant)) | |
| 5 | 53 | 49 | 55 | √ | × | − | Bone, liver | Avastin |
| 6 | 40 | 33 | 41 | √ | × | − | Bone, liver | Avastin and Chemotherapy (cisplatin/gemzar) |
| 7 | 33 | 33 | √ | × | + | Local disease | Targeted therapy for HER2 and Chemotherapy (navalbine (neaoadjuvant)) | |
| 8 | 64 | 54 | 66 | √ | × | − | Bone, liver | Chemotherapy (carboplatin/gemzar) |
| 9 | 38 | 32 | 39 | √ | × | + | Bone, lung | Targeted therapy for HER2 |
| 10 | 37 | 36 | √ | × | + | Brain | Targeted therapy for HER2 | |
| 11 | 62 | 46 | 64 | √ | × | − | Liver, bone | Chemotherapy (carboplatin/gemzar) |
| 12 | 84 | 76 | √ | × | + | Local disease | Targeted therapy for HER2 and Chemotherapy (navalbine (neaoadjuvant)) | |
| 13 | 54 | 36 | 56 | √ | × | − | Bone, lung, liver | Chemotherapy (cisplatin/leucovorin/5FU 3) |
| 14 | 58 | 58 | √ | × | + | Targeted therapy for HER2 | ||
| 15 | 71 | 69 | 72 | √ | × | − | Lung, liver, bone | Hormonal |
| 16 | 53 | 53 | √ | × | − | Chemotherapy (taxol (adjuvant)) | ||
| 17 | 55 | 49 | √ | × | − | Lung, bone | Avastin and Chemotherapy (navalbine) | |
| 18 | 85 | 80 | 85 | √ | × | − | Liver, bone, lung | Chemotherapy (carboplatin) |
| 19 | 49 | 48 | √ | × | + | Targeted therapy for HER2 | ||
| 20 | 35 | 35 | √ | × | − | Local disease | Chemotherapy (taxol (neoadjuvant)) | |
| 21 | 67 | 64 | 68 | × | √ | − | Brain, bone, lung | Chemotherapy (carboplatin + taxol) |
| 22 | 47 | 42 | 47 | √ | × | − | Bone, liver | Chemotherapy (carboplatin/gemzar) |
| 23 | 46 | 45 | √ | × | + | Targeted therapy for HER2 | ||
| 24 | 54 | 41 | 55 | √ | × | − | Bone | Hormonal and Palbocyclib |
| 25 | 51 | 51 | 52 | √ | × | − | Bone | Hormonal and Palbocyclib |
| 26 | 35 | 29 | 36 | √ | × | − | Liver, bone, adrenal gland | Hormonal and Afinitor |
| 27 | 62 | 62 | × | √ | − | Chemotherapy (taxol (adjuvant)) | ||
| 28 | 39 | 39 | 40 | √ | × | − | Bone | Hormonal and Palbocyclib |
| 29 | 58 | 52 | 59 | √ | × | − | Bone | Chemotherapy (taxol) |
| 30 | 56 | 43 | 57 | √ | × | − | Liver, bone | Hormonal and Afinitor |
| 31 | 52 | 52 | 53 | √ | × | − | Bone, liver | Chemotherapy (xeloda) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bentata, M.; Morgenstern, G.; Nevo, Y.; Kay, G.; Granit Mizrahi, A.; Temper, M.; Maimon, O.; Monas, L.; Basheer, R.; Ben-Hur, A.; et al. Splicing Factor Transcript Abundance in Saliva as a Diagnostic Tool for Breast Cancer. Genes 2020, 11, 880. https://doi.org/10.3390/genes11080880
Bentata M, Morgenstern G, Nevo Y, Kay G, Granit Mizrahi A, Temper M, Maimon O, Monas L, Basheer R, Ben-Hur A, et al. Splicing Factor Transcript Abundance in Saliva as a Diagnostic Tool for Breast Cancer. Genes. 2020; 11(8):880. https://doi.org/10.3390/genes11080880
Chicago/Turabian StyleBentata, Mercedes, Guy Morgenstern, Yuval Nevo, Gillian Kay, Avital Granit Mizrahi, Mark Temper, Ofra Maimon, Liza Monas, Reham Basheer, Asa Ben-Hur, and et al. 2020. "Splicing Factor Transcript Abundance in Saliva as a Diagnostic Tool for Breast Cancer" Genes 11, no. 8: 880. https://doi.org/10.3390/genes11080880
APA StyleBentata, M., Morgenstern, G., Nevo, Y., Kay, G., Granit Mizrahi, A., Temper, M., Maimon, O., Monas, L., Basheer, R., Ben-Hur, A., Peretz, T., & Salton, M. (2020). Splicing Factor Transcript Abundance in Saliva as a Diagnostic Tool for Breast Cancer. Genes, 11(8), 880. https://doi.org/10.3390/genes11080880