Co-Expression Networks for Causal Gene Identification Based on RNA-Seq Data of Corynebacterium pseudotuberculosis

 , , , , , ,
, , , , , ,  , and
, and
Abstract
1. Introduction
2. Methodology
2.1. Bacterial Strains and Growth Conditions
2.2. Expression Datasets
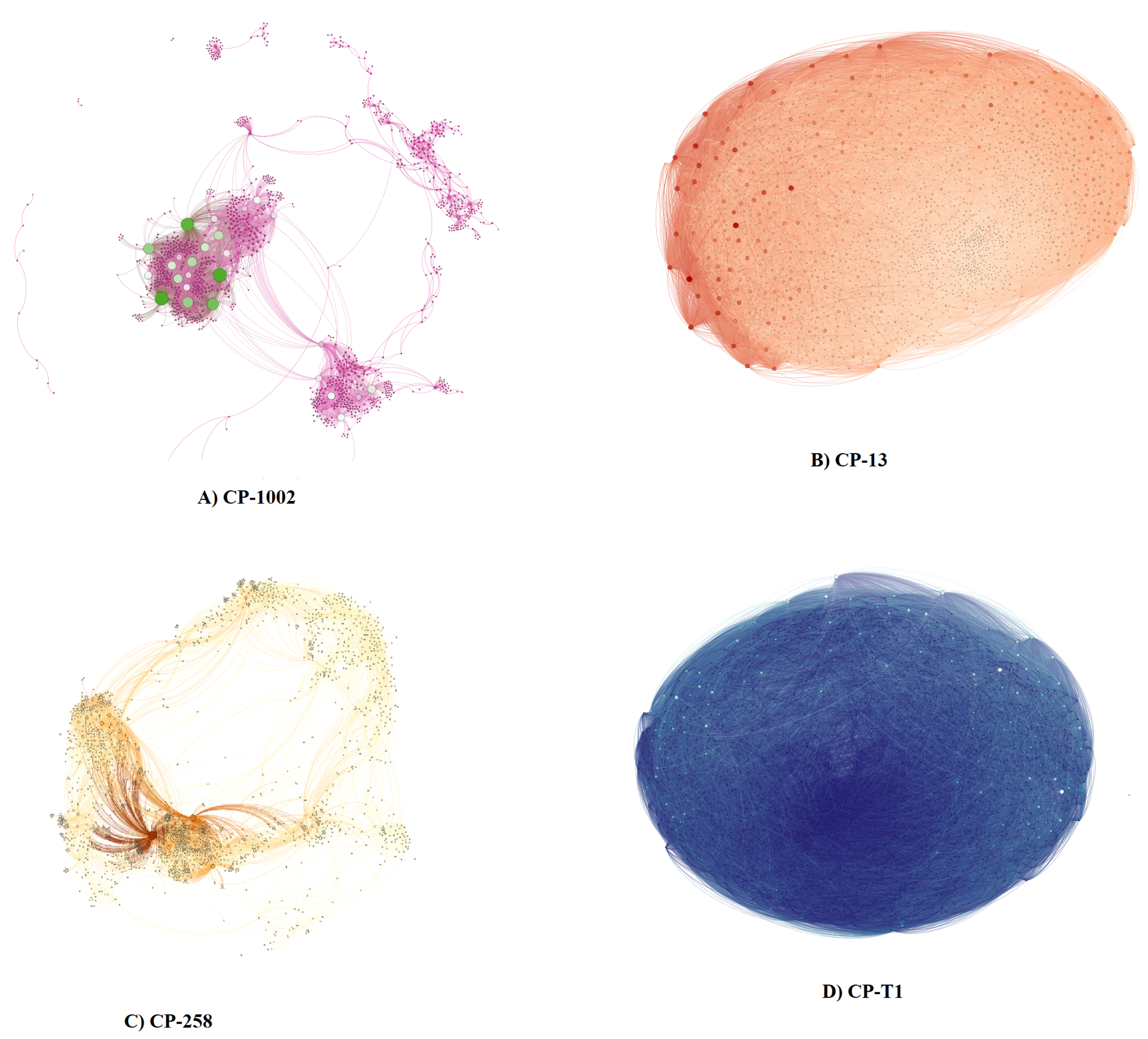
2.3. Network Analysis
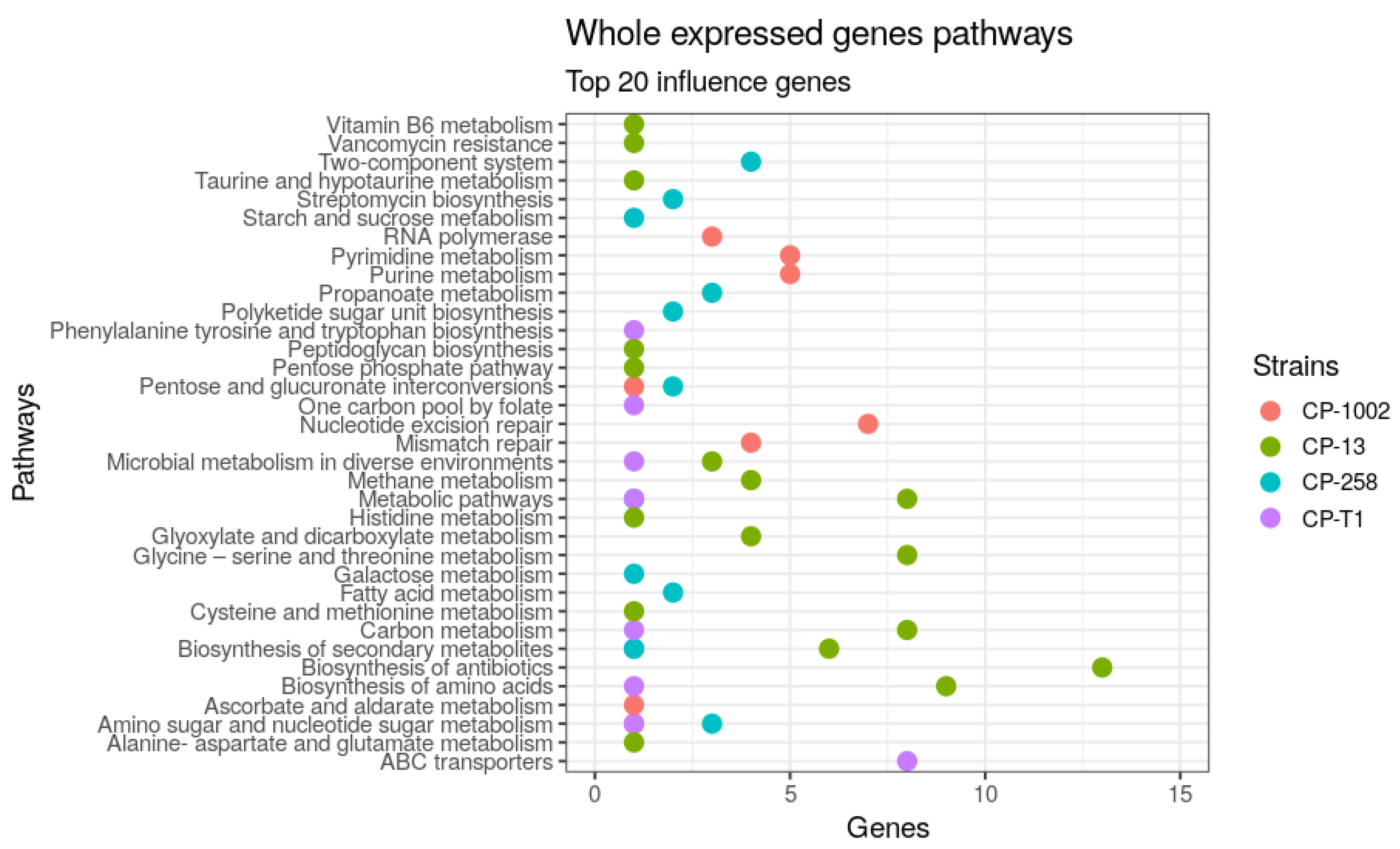
2.4. Identification of Causal Genes
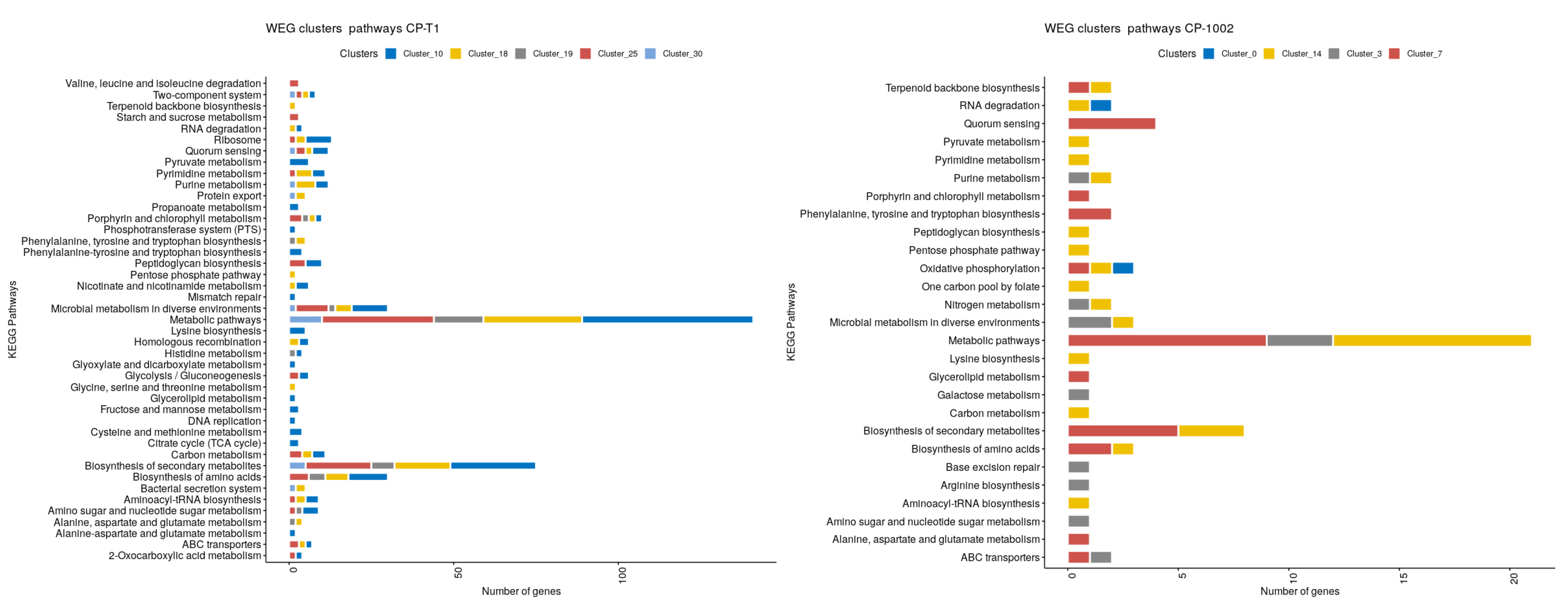
2.5. Network Clustering
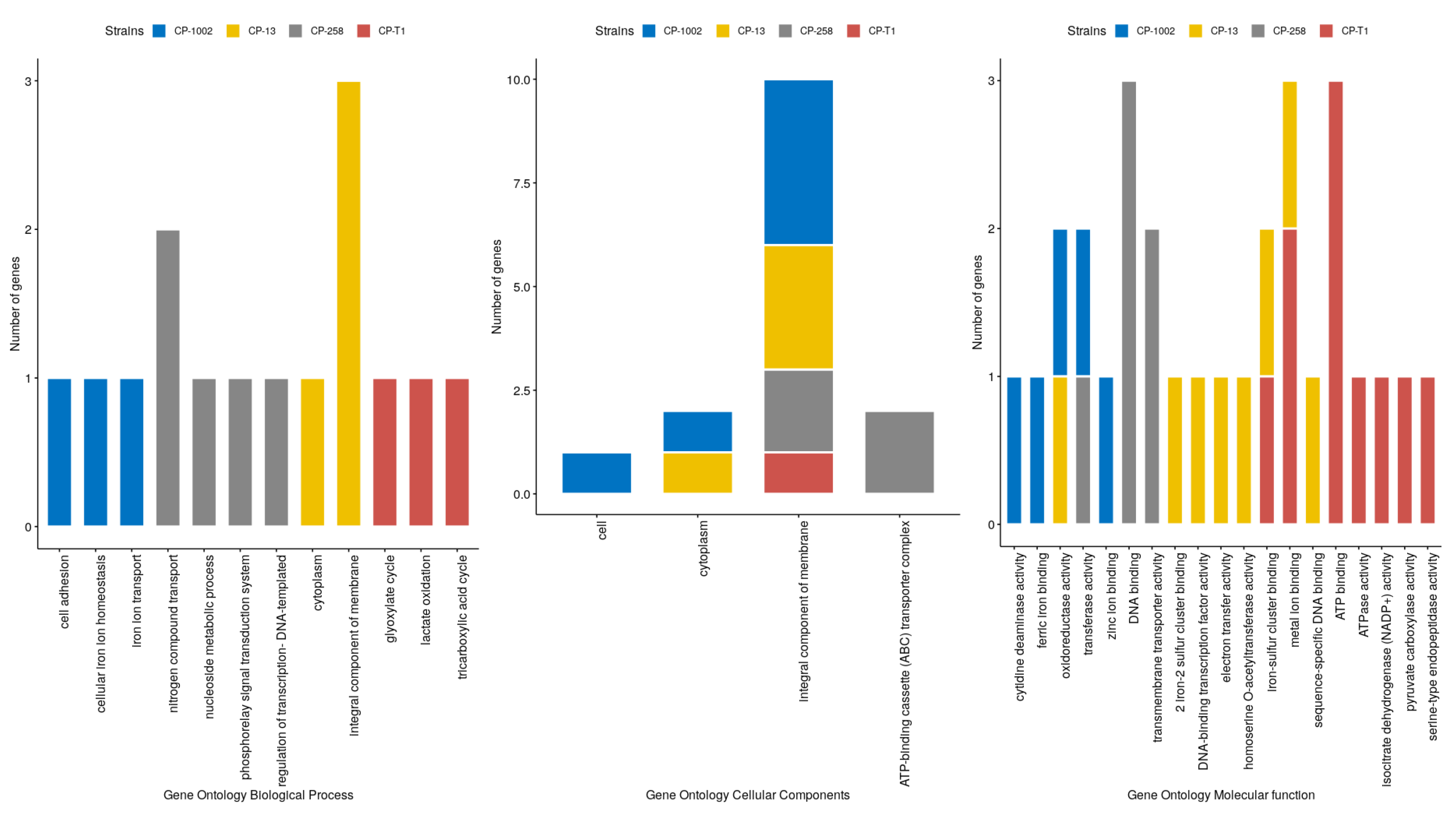
2.6. Sub-Network Detection and Enrichment Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CEM | Co-expression networks |
| TPM | Transcripts per kilobase million |
| WEG | Whole genome gene |
| DE | Differentially expressed genes |
References
- Silva, W.M.; Dorella, F.A.; Soares, S.C.; Souza, G.H.; Castro, T.L.; Seyffert, N.; Figueiredo, H.; Miyoshi, A.; Le Loir, Y.; Silva, A.; et al. A shift in the virulence potential of Corynebacterium pseudotuberculosis biovar ovis after passage in a murine host demonstrated through comparative proteomics. BMC Microbiol. 2017, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- De Sá, P.H.; Veras, A.A.; Carneiro, A.R.; Baraúna, R.A.; Guimarães, L.C.; Pinheiro, K.C.; Pinto, A.C.; Soares, S.C.; Schneider, M.P.; Azevedo, V.; et al. Corynebacterium pseudotuberculosis RNA-Seq data from abiotic stresses. Data Brief 2015, 5, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.C.; de Sá, P.H.C.G.; Ramos, R.T.; Barbosa, S.; Barbosa, H.P.M.; Ribeiro, A.C.; Silva, W.M.; Rocha, F.S.; Santana, M.P.; de Paula Castro, T.L.; et al. Differential transcriptional profile of Corynebacterium pseudotuberculosis in response to abiotic stresses. BMC Genom. 2014, 15, 14. [Google Scholar] [CrossRef] [PubMed]
- Silva, W.M.; Seyffert, N.; Santos, A.V.; Castro, T.L.; Pacheco, L.G.; Santos, A.R.; Ciprandi, A.; Dorella, F.A.; Andrade, H.M.; Barh, D.; et al. Identification of 11 new exoproteins in Corynebacterium pseudotuberculosis by comparative analysis of the exoproteome. Microb. Pathog. 2013, 61, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Santana-Jorge, K.T.; Santos, T.M.; Tartaglia, N.R.; Aguiar, E.L.; Souza, R.F.; Mariutti, R.B.; Eberle, R.J.; Arni, R.K.; Portela, R.W.; Meyer, R.; et al. Putative virulence factors of Corynebacterium pseudotuberculosis FRC41: Vaccine potential and protein expression. Microb. Cell Factories 2016, 15, 83. [Google Scholar] [CrossRef]
- Cerdeira, L.T.; Schneider, M.P.C.; Pinto, A.C.; de Almeida, S.S.; dos Santos, A.R.; Barbosa, E.G.V.; Ali, A.; Aburjaile, F.F.; de Abreu, V.A.C.; Guimarães, L.C.; et al. Complete Genome Sequence of Corynebacterium pseudotuberculosis Strain CIP 52.97, Isolated from a Horse in Kenya. J. Bacteriol. 2011, 193, 7025–7026. [Google Scholar] [CrossRef]
- Barh, D.; Gupta, K.; Jain, N.; Khatri, G.; León-Sicairos, N.; Canizalez-Roman, A.; Tiwari, S.; Verma, A.; Rahangdale, S.; Shah Hassan, S.; et al. Conserved host–pathogen PPIs Globally conserved inter-species bacterial PPIs based conserved host-pathogen interactome derived novel target in C. pseudotuberculosis, C. diphtheriae, M. tuberculosis, C. ulcerans, Y. pestis, and E. coli targeted by Piper betel compounds. Integr. Biol. 2013, 5, 495–509. [Google Scholar] [CrossRef]
- Morales, N.; Aldridge, D.; Bahamonde, A.; Cerda, J.; Araya, C.; Muñoz, R.; Saldías, M.E.; Lecocq, C.; Fresno, M.; Abalos, P.; et al. Corynebacterium pseudotuberculosis infection in Patagonian Huemul (Hippocamelus bisulcus). J. Wildl. Dis. 2017, 53, 621–624. [Google Scholar] [CrossRef]
- Chaitankar, V.; Ghosh, P.; Perkins, E.; Gong, P.; Zhang, C. Time lagged information theoretic approaches to the reverse engineering of gene regulatory networks. BMC Bioinform. 2010, 11, S19. [Google Scholar] [CrossRef]
- Chaitankar, V.; Ghosh, P.; Perkins, E.; Gong, P.; Deng, Y.; Zhang, C. A novel gene network inference algorithm using predictive minimum description length approach. BMC Syst. Biol. 2010, 4, S7. [Google Scholar] [CrossRef] [PubMed]
- Chai, L.E.; Loh, S.K.; Low, S.T.; Mohamad, M.S.; Deris, S.; Zakaria, Z. A review on the computational approaches for gene regulatory network construction. Comput. Biol. Med. 2014, 48, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Marbach, D.; Costello, J.C.; Küffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; Aderhold, A.; Bonneau, R.; Chen, Y.; et al. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796. [Google Scholar] [CrossRef] [PubMed]
- Tieri, P.; Farina, L.; Petti, M.; Astolfi, L.; Paci, P.; Castiglione, F. Network Inference and Reconstruction in Bioinformatics. In Encyclopedia of Bioinformatics and Computational Biology; Ranganathan, S., Gribskov, M., Nakai, K., Schönbach, C., Eds.; Academic Press: Oxford, UK, 2019; pp. 805–813. [Google Scholar] [CrossRef]
- Suratanee, A.; Chokrathok, C.; Chutimanukul, P.; Khrueasan, N.; Buaboocha, T.; Chadchawan, S.; Plaimas, K. Two-State Co-Expression Network Analysis to Identify Genes Related to Salt Tolerance in Thai Rice. Genes 2018, 9, 594. [Google Scholar] [CrossRef] [PubMed]
- Nalluri, J.J.; Rana, P.; Barh, D.; Azevedo, V.; Dinh, T.N.; Vladimirov, V.; Ghosh, P. Determining causal miRNAs and their signaling cascade in diseases using an influence diffusion model. Sci. Rep. 2017, 7, 8133. [Google Scholar] [CrossRef] [PubMed]
- Guille, A.; Hacid, H.; Favre, C.; Zighed, D.A. Information diffusion in online social networks: A survey. ACM Sigmod Rec. 2013, 42, 17–28. [Google Scholar] [CrossRef]
- Nalluri, J.J.; Barh, D.; Azevedo, V.; Ghosh, P. miRsig: A consensus-based network inference methodology to identify pan-cancer miRNA-miRNA interaction signatures. Sci. Rep. 2017, 7, 39684. [Google Scholar] [CrossRef]
- Ruiz, J.C.; D’Afonseca, V.; Silva, A.; Ali, A.; Pinto, A.C.; Santos, A.R.; Rocha, A.A.; Lopes, D.O.; Dorella, F.A.; Pacheco, L.G.; et al. Evidence for reductive genome evolution and lateral acquisition of virulence functions in two Corynebacterium pseudotuberculosis strains. PLoS ONE 2011, 6, e18551. [Google Scholar] [CrossRef]
- Gomide, A.C.P.; de Sa, P.G.; Cavalcante, A.L.Q.; de Jesus Sousa, T.; Gomes, L.G.R.; Ramos, R.T.J.; Azevedo, V.; Silva, A.; Folador, A.R.C. Heat shock stress: Profile of differential expression in Corynebacterium pseudotuberculosis biovar Equi. Gene 2018, 645, 124–130. [Google Scholar] [CrossRef]
- Dorella, F.A.; Estevam, E.M.; Pacheco, L.G.; Guimarães, C.T.; Lana, U.G.; Gomes, E.A.; Barsante, M.M.; Oliveira, S.C.; Meyer, R.; Miyoshi, A.; et al. In vivo insertional mutagenesis in Corynebacterium pseudotuberculosis: An efficient means to identify DNA sequences encoding exported proteins. Appl. Environ. Microbiol. 2006, 72, 7368–7372. [Google Scholar] [CrossRef]
- Ribeiro, D.; de Souza Rocha, F.; Leite, K.M.C.; de Castro Soares, S.; Silva, A.; Portela, R.W.D.; Meyer, R.; Miyoshi, A.; Oliveira, S.C.; Azevedo, V.; et al. An iron-acquisition-deficient mutant of Corynebacterium pseudotuberculosis efficiently protects mice against challenge. Vet. Res. 2014, 45, 28. [Google Scholar] [CrossRef][Green Version]
- Ibraim, I.C.; Parise, M.T.D.; Parise, D.; Sfeir, M.Z.T.; de Paula Castro, T.L.; Wattam, A.R.; Ghosh, P.; Barh, D.; Souza, E.M.; Góes-Neto, A.; et al. Transcriptome profile of Corynebacterium pseudotuberculosis in response to iron limitation. BMC Genom. 2019, 20, 663. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 February 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-Seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2007, 36, D13–D21. [Google Scholar] [CrossRef]
- Markowitz, V.M.; Chen, I.M.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Grechkin, Y.; Ratner, A.; Jacob, B.; Huang, J.; Williams, P.; et al. IMG: The integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012, 40, D115–D122. [Google Scholar] [CrossRef]
- Feng, J.; Meyer, C.A.; Wang, Q.; Liu, J.S.; Shirley Liu, X.; Zhang, Y. GFOLD: A generalized fold change for ranking differentially expressed genes from RNA-Seq data. Bioinformatics 2012, 28, 2782–2788. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Warden, C.D.; Yuan, Y.C.; Wu, X. Optimal calculation of RNA-Seq fold-change values. Int. J. Comput. Bioinform. Silico Model. 2013, 2, 285–292. [Google Scholar]
- Chen, W.H.; Lu, G.; Chen, X.; Zhao, X.M.; Bork, P. OGEE v2: An update of the online gene essentiality database with special focus on differentially essential genes in human cancer cell lines. Nucleic Acids Res. 2017, 45, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Rocha, D.J.; Santos, C.S.; Pacheco, L.G. Bacterial reference genes for gene expression studies by RT-qPCR: Survey and analysis. Antonie Van Leeuwenhoek 2015, 108, 685–693. [Google Scholar] [CrossRef] [PubMed]
- MacQueen, J. Some methods for classification and analysis of multivariate observations. In Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability, Oakland, CA, USA, 27 December 1967; Volume 1, pp. 281–297. [Google Scholar]
- Pavlopoulos, G.A.; Secrier, M.; Moschopoulos, C.N.; Soldatos, T.G.; Kossida, S.; Aerts, J.; Schneider, R.; Bagos, P.G. Using graph theory to analyze biological networks. BioData Min. 2011, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.H.; Apeltsin, L.; Newman, A.M.; Baumbach, J.; Wittkop, T.; Su, G.; Bader, G.D.; Ferrin, T.E. clusterMaker: A multi-algorithm clustering plugin for Cytoscape. BMC Bioinform. 2011, 12, 436. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Araujo, F.A.; Barh, D.; Silva, A.; Guimarães, L.; Ramos, R.T.J. GO FEAT: A rapid web-based functional annotation tool for genomic and transcriptomic data. Sci. Rep. 2018, 8, 1794. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape stringApp: Network analysis and visualization of proteomics data. J. Proteome Res. 2018, 18, 623–632. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics: A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin, Germany, 2016. [Google Scholar]
- Kassambara, A. ggpubr:“ggplot2” Based Publication Ready Plots. R Package Version 0.1 2017, 6. Available online: https://rpkgs.datanovia.com/ggpubr/ (accessed on 12 February 2019).
- Radkov, A.D.; Hsu, Y.P.; Booher, G.; VanNieuwenhze, M.S. Imaging bacterial cell wall biosynthesis. Annu. Rev. Biochem. 2018, 87, 991–1014. [Google Scholar] [CrossRef] [PubMed]
- McKean, S.; Davies, J.; Moore, R. Identification of macrophage induced genes of Corynebacterium pseudotuberculosis by differential fluorescence induction. Microbes Infect. 2005, 7, 1352–1363. [Google Scholar] [CrossRef] [PubMed]
- Merkamm, M.; Guyonvarch, A. Cloning of the sodA Gene fromCorynebacterium melassecola and Role of Superoxide Dismutase in Cellular Viability. J. Bacteriol. 2001, 183, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- den Hengst, C.D.; Buttner, M.J. Redox control in actinobacteria. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2008, 1780, 1201–1216. [Google Scholar] [CrossRef] [PubMed]
- Newton, G.L.; Av-Gay, Y.; Fahey, R.C. A novel mycothiol-dependent detoxification pathway in mycobacteria involving mycothiol S-conjugate amidase. Biochemistry 2000, 39, 10739–10746. [Google Scholar] [CrossRef] [PubMed]
- Rawat, M.; Newton, G.L.; Ko, M.; Martinez, G.J.; Fahey, R.C.; Av-Gay, Y. Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob. Agents Chemother. 2002, 46, 3348–3355. [Google Scholar] [CrossRef]
- Seebeck, F.P. In vitro reconstitution of Mycobacterial ergothioneine biosynthesis. J. Am. Chem. Soc. 2010, 132, 6632–6633. [Google Scholar] [CrossRef]
- Cheah, I.K.; Halliwell, B. Ergothioneine; antioxidant potential, physiological function and role in disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 784–793. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Satorhelyi, P. Microarray-Analyse der pH-Stressantwort von Listeria Monocytogenes und Corynebacterium Glutamicum. Ph.D. Thesis, Technische Universität München, München, Germany, 2005. [Google Scholar]
- Weeks, D.L.; Sachs, G. Sites of pH regulation of the urea channel of Helicobacter pylori. Mol. Microbiol. 2001, 40, 1249–1259. [Google Scholar] [CrossRef]
- Hanna, M.N.; Ferguson, R.J.; Li, Y.H.; Cvitkovitch, D.G. uvrA is an acid-inducible gene involved in the adaptive response to low pH inStreptococcus mutans. J. Bacteriol. 2001, 183, 5964–5973. [Google Scholar] [CrossRef]
- Heermann, R.; Jung, K. Structural features and mechanisms for sensing high osmolarity in microorganisms. Curr. Opin. Microbiol. 2004, 7, 168–174. [Google Scholar] [CrossRef]
- Dodson, K.W.; Pinkner, J.S.; Rose, T.; Magnusson, G.; Hultgren, S.J.; Waksman, G. Structural basis of the interaction of the pyelonephritic E. coli adhesin to its human kidney receptor. Cell 2001, 105, 733–743. [Google Scholar] [CrossRef]
- Park, J.H.; Roe, J.H. Mycothiol regulates and is regulated by a thiol-specific antisigma factor RsrA and σR in Streptomyces coelicolor. Mol. Microbiol. 2008, 68, 861–870. [Google Scholar] [CrossRef]
- Martín, J.F. Phosphate control of the biosynthesis of antibiotics and other secondary metabolites is mediated by the PhoR-PhoP system: An unfinished story. J. Bacteriol. 2004, 186, 5197–5201. [Google Scholar] [CrossRef]
- Liu, G.; Chater, K.F.; Chandra, G.; Niu, G.; Tan, H. Molecular regulation of antibiotic biosynthesis in Streptomyces. Microbiol. Mol. Biol. Rev. 2013, 77, 112–143. [Google Scholar] [CrossRef] [PubMed]
- Bromke, M.A. Amino acid biosynthesis pathways in diatoms. Metabolites 2013, 3, 294–311. [Google Scholar] [CrossRef]
- Kisker, C.; Kuper, J.; Van Houten, B. Prokaryotic nucleotide excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012591. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Size (Mb) | GC% | Protein | Genes |
|---|---|---|---|---|
| CP-13 | 2.34 | 52.2 | 2013 | 2135 |
| CP-T1 | 2.34 | 52.2 | 2008 | 2125 |
| CP-258 | 2.37 | 52.1 | 2038 | 2165 |
| CP-1002B | 2.34 | 52.2 | 2009 | 2124 |
| Strains 1 | Strains 2 | ANI% |
|---|---|---|
| CP-13 | CP-258 | 98.9036 |
| CP-13 | CP-1002B | 99.9966 |
| CP-1002B | CP-258 | 98.9051 |
| CP-1002B | CP-T1 | 99.9968 |
| CP-T1 | CP-258 | 98.904 |
| CP-T1 | CP-13 | 100 |
| Strains | CP-13 | CP-258 | CP-1002 | CP-T1 |
|---|---|---|---|---|
| Whole Genomes | 2113 | 2064 | 2091 | 2093 |
| Differentially Expressed Genes | 63 | 139 | 168 | 93 |
| Genes Cp-13 | Product | Genes Cp-258 | Product | Genes Cp-1002 | Product | Genes Cp-T1 | Product |
|---|---|---|---|---|---|---|---|
| mca | Mycothiol S-conjugate amidase | CP258_RS02980 | Methylmalonyl-CoA carboxyltransferase 12S subunit | Cp1002B_RS03590 | Hypothetical protein | pknL | Serine/threonine protein kinase |
| hisN | Histidinol-phosphatase | CP258_RS03105 | LPxTG domain-containing protein | mcbR | TetR family transcriptional regulator | fhs | Formate–tetrahydrofolate ligase |
| rbsK | Ribokinase | CP258_RS03110 | Aminotransferase | ureE | Urease accessory protein UreE | ogt | Methylated-DNA–protein-cysteine methyltransferase |
| pepC2 | M18 family aminopeptidase | tcsS4 | Two component system sensor kinase protein | uvrD3 | DNA helicase | Cp13_RS09405 | Oxidoreductase |
| tcsR3 | Two-component system transcriptional regulatory protein | CP258_RS04420 | Hypothetical protein | Cp1002B_RS02755 | Abi family protein | pbpB | Penicillin binding protein transpeptidase |
| Cp13_RS10225 | Antimicrobial peptide ABC transporter ATPase | rimJ | Ribosomal-protein-alanine acetyltransferase | cmtB | Trehalose corynomycolyl transferase B | nodI | Nod factor export ATP- binding protein I |
| ald | Alanine dehydrogenase | CP258_RS03575 | ABC transporter ATP-binding protein | whiB | Transcriptional regulator WhiB | Cp13_RS04005 | Hypothetical protein |
| mrpD | Na(+)/H(+) antiporter subunit D | tehA | C4-dicarboxylate transporter/malic acid transport protein | rplO | 50S ribosomal protein L15 | pafA2 | Pup–protein ligase |
| serC | Phosphoserine transaminase | CP258_RS03580 | Antibiotic biosynthesis monooxygenase | Cp1002B_RS02920 | LPxTG domain-containing protein | Cp13_RS01180 | Secreted hydrolase |
| mgtA | Glycosyl transferase group 1 | echA6 | Enoyl-CoA hydratase echA6 | rluC | Ribosomal pseudouridine synthase | trpC | N-(5’-phosphoribosyl)anthranilate isomerase |
| Cp13_RS01600 | ABC-type metal ion transport system, periplasmic component/surface adhesin | rmlD | dTDP-4-dehydrorhamnose reductase | yhcL | Cryptic C4-dicarboxylate membrane transporter dcuD | Cp13_RS08505 | Acetyltransferase |
| sdaA | L-serine dehydratase | CP258_RS07545 | Hypothetical protein | Cp1002B_RS01655 | ABC transporter inner membrane protein | nagB | Glucosamine-6-phosphate deaminase |
| Cp13_RS07170 | Glyoxalase/bleomycin resistance protein/dioxygenase | galU | UTP–glucose-1-phosphate uridylyltransferase | Cp1002B_RS10840 | LPxTG domain-containing protein | Cp13_RS03565 | Hypothetical protein |
| Cp13_RS01385 | Hypothetical protein | cstA | Response regulator | Cp1002B_RS01705 | Hypothetical protein | Cp13_RS05420 | ABC transporter ATP-binding protein |
| lpdA | Flavoprotein disulfide reductase | argS | Arginine–tRNA ligase | Cp1002B_RS10170 | ABC transporter | yvrC | ABC transporter substrate-binding protein |
| mraY | Phospho-N-acetylmuramoyl pentapeptide-transferase | hpf | Ribosome hibernation promoting factor | fadF | Protein fadF | fagA | Hypothetical protein |
| Cp13_RS08655 | Hemolysin III-like protein | deoA | Thymidine phosphorylase | udgA | UDP-glucose 6-dehydrogenase | mnmA | tRNA-specific 2-thiouridylase MnmA |
| copD | Copper resistance D domain- containing protein/Cytochrome c oxidase caa3 assembly factor (Caa3_CtaG) | mprA_2 | Two component system response regulator | gltT | Sodium/glutamate symporter | Cp13_RS06640 | Hypothetical protein |
| glmS | Glutamine–fructose-6-phosphate aminotransferase [isomerizing] | oppC2 | Oligopeptide transport system permease OppC | Cp1002B_RS01200 | Serine proteases of the peptidase family S9A | Cp13_RS07860 | Phosphoglycerate dehydrogenase |
| Cp13_RS10090 | NYN domain-containing protein | scpB | Segregation and condensation protein B | uvrA | UvrABC system protein A | Cp13_RS03800 | Putative secreted protein |
| Genes Cp-13 | Product | Genes Cp-258 | Product | Genes Cp-1002 | Product | Genes Cp-T1 | Product |
|---|---|---|---|---|---|---|---|
| desA3 | Stearoyl-CoA 9-desaturase | CP258_RS02710 | Putative secreted protein | cynT | Carbonic anhydrase | pyc | Pyruvate carboxylase |
| htaA | Cell-surface hemin receptor | CP258_RS02905 | Hypothetical protein | Cp1002B_RS03070 | HtaA domain-containing protein | fecE | Fe(3+) dicitrate transport ATP-binding protein FecE |
| Cp13_RS02285 | Hypothetical protein | vapI | Virulence-associated protein | Cp1002B_RS02920 | LPxTG domain-containing protein | hmuT | Hemin-binding periplasmic protein |
| lutB | Putative iron-sulfur protein | tetR3 | TetR family transcriptional regulator | cdd | Cytidine deaminase | Cp13_RS04105 | LUD_dom domain-containing protein |
| Cp13_RS04105 | LUD_dom domain-containing protein | rshA | Anti-sigma factor | Cp1002B_RS03075 | Hypothetical protein | Cp13_RS02285 | Hypothetical protein |
| htaB | Cell-surface hemin receptor | cstA | Response regulator | Cp1002B_RS03455 | Oxidoreductase | lysA2 | Diaminopimelate decarboxylase |
| Cp13_RS02610 | Stearoyl-CoA 9-desaturase electron transfer partner | gluC | Glutamate transport system permease protein gluC | Cp1002B_RS03180 | Hypothetical protein | icd | Isocitrate dehydrogenase [NADP] |
| Cp13_RS09955 | Flavin reductase | odhI | Oxoglutarate dehydrogenase inhibitor | ftn | Ferritin | sprT | Trypsin |
| metX | Homoserine O-acetyltransferase | yecS | ABC transporter domain-containing protein | dnaK | Chaperone protein DnaK | gluA | Glutamine ABC transporter ATP-binding protein |
| ripA_2 | HTH-type transcriptional repressor of iron protein A | ykoE | HMP/thiamine permease protein ykoE | glpQ_1 | Glycerophosphoryl diester phosphodiesterase | lutB | Putative iron-sulfur protein |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, E.F.; Rana, P.; Queiroz Cavalcante, A.L.; da Silva, A.L.; Cybelle Pinto Gomide, A.; Carneiro Folador, A.R.; Azevedo, V.; Ghosh, P.; T. J. Ramos, R. Co-Expression Networks for Causal Gene Identification Based on RNA-Seq Data of Corynebacterium pseudotuberculosis. Genes 2020, 11, 794. https://doi.org/10.3390/genes11070794
Franco EF, Rana P, Queiroz Cavalcante AL, da Silva AL, Cybelle Pinto Gomide A, Carneiro Folador AR, Azevedo V, Ghosh P, T. J. Ramos R. Co-Expression Networks for Causal Gene Identification Based on RNA-Seq Data of Corynebacterium pseudotuberculosis. Genes. 2020; 11(7):794. https://doi.org/10.3390/genes11070794
Chicago/Turabian StyleFranco, Edian F., Pratip Rana, Ana Lidia Queiroz Cavalcante, Artur Luiz da Silva, Anne Cybelle Pinto Gomide, Adriana R. Carneiro Folador, Vasco Azevedo, Preetam Ghosh, and Rommel T. J. Ramos. 2020. "Co-Expression Networks for Causal Gene Identification Based on RNA-Seq Data of Corynebacterium pseudotuberculosis" Genes 11, no. 7: 794. https://doi.org/10.3390/genes11070794
APA StyleFranco, E. F., Rana, P., Queiroz Cavalcante, A. L., da Silva, A. L., Cybelle Pinto Gomide, A., Carneiro Folador, A. R., Azevedo, V., Ghosh, P., & T. J. Ramos, R. (2020). Co-Expression Networks for Causal Gene Identification Based on RNA-Seq Data of Corynebacterium pseudotuberculosis. Genes, 11(7), 794. https://doi.org/10.3390/genes11070794