Clinical and Molecular Data Define a Diagnosis of Arrhythmogenic Cardiomyopathy in a Carrier of a Brugada-Syndrome-Associated PKP2 Mutation



 , , , ,
, , , ,
Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Genotype Analysis
2.3. Cardiac Mesenchymal Stromal Cell isolation
2.4. Oil Red O Staining
2.5. Real-Time PCR
2.6. Western Blot Analysis
2.7. Immunofluorescence
2.8. Statistical Analysis
3. Results
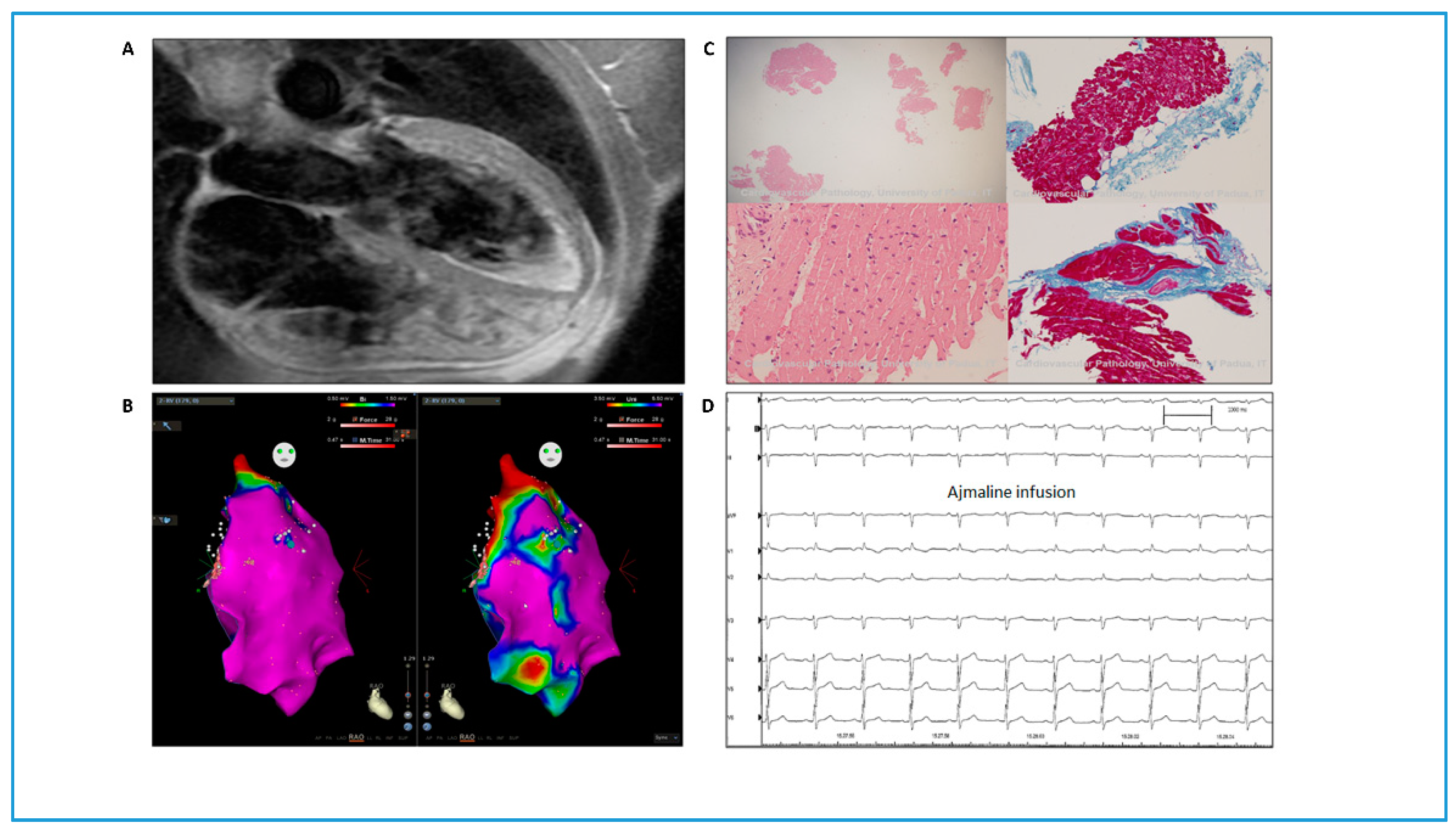
3.1. Clinical Data
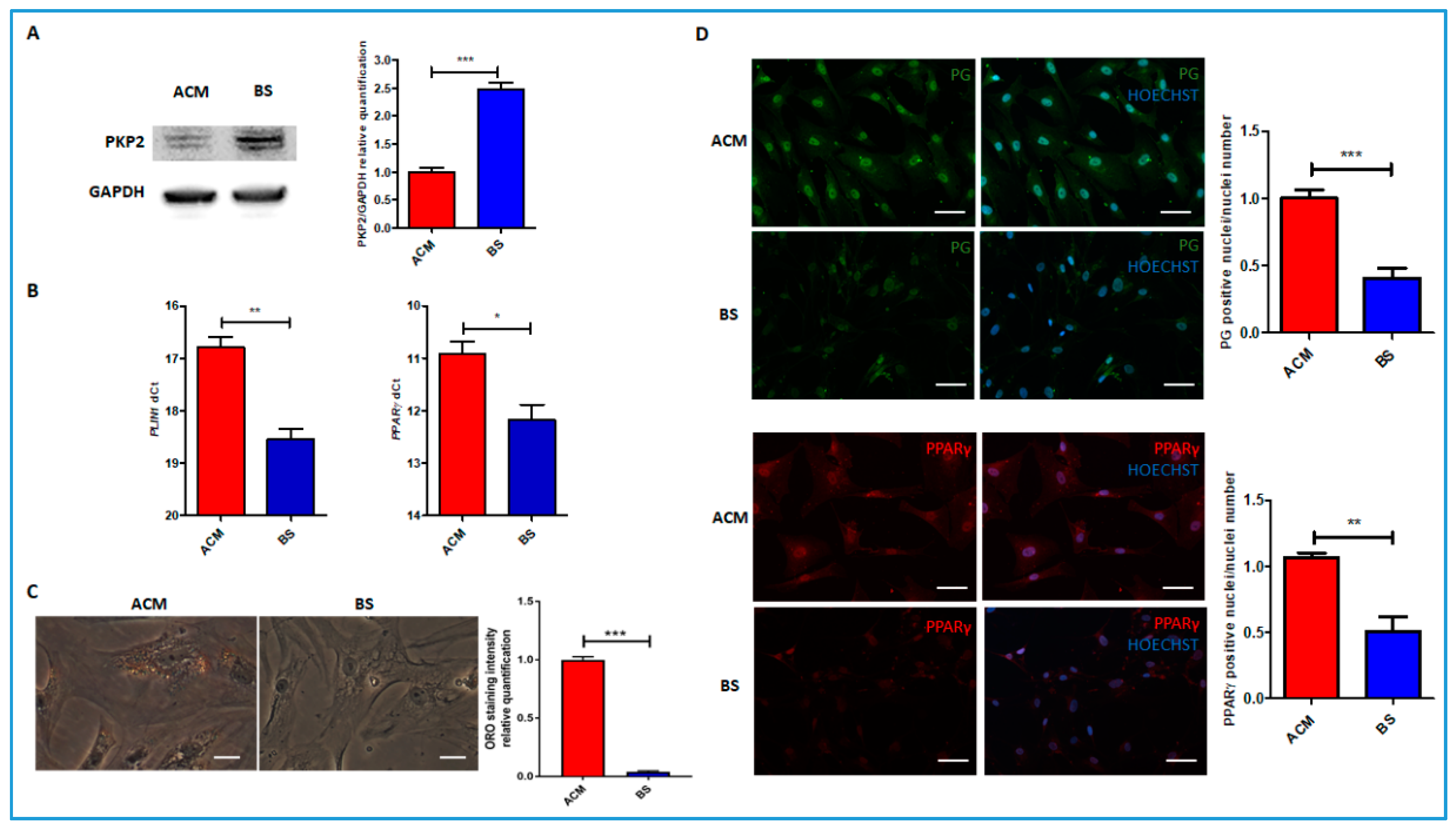
3.2. Molecular Data
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Tintelen, J.P.; Entius, M.M.; A Bhuiyan, Z.; Jongbloed, R.; Wiesfeld, A.C.; Wilde, A.A.M.; Van Der Smagt, J.; Boven, L.G.; Mannens, M.; Van Langen, I.M.; et al. Plakophilin-2 Mutations Are the Major Determinant of Familial Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circulation 2006, 113, 1650–1658. [Google Scholar] [CrossRef] [PubMed]
- Sieira, J.; Dendramis, G.; Brugada, P. Pathogenesis and management of Brugada syndrome. Nat. Rev. Cardiol. 2016, 13, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Minamino, T. Genetics of Brugada syndrome. J. Hum. Genet. 2015, 61, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Tavora, F.; Oliveira, J.; Li, L.; Franco, M.; Fowler, D.; Zhao, Z.; Burke, A. PKP2 mutations in sudden death from arrhythmogenic right ventricular cardiomyopathy (ARVC) and sudden unexpected death with negative autopsy (SUDNA). Circ. J. 2011, 76, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Catalano, O.; Antonaci, S.; Moro, G.; Mussida, M.; Frascaroli, M.; Baldi, M.; Cobelli, F.; Baiardi, P.; Nastoli, J.; Bloise, R.; et al. Magnetic resonance investigations in Brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur. Hear. J. 2009, 30, 2241–2248. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Delmar, M. Desmosomes and the sodium channel complex: Implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends Cardiovasc. Med. 2014, 24, 184–190. [Google Scholar] [CrossRef]
- Riele, A.S.T.; Agullo-Pascual, E.; A James, C.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Rothenberg, E.; Sobreira, N.L.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Gusky, H.C.; Novelli, V.; Kim, C.; Tirasawasdichai, T.; Judge, D.P.; et al. Missense Mutations in Plakophilin-2 Cause Sodium Current Deficit and Associate with a Brugada Syndrome Phenotype. Circulation 2013, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Sommariva, E.; Brambilla, S.; Carbucicchio, C.; Gambini, E.; Meraviglia, V.; Russo, A.D.; Farina, F.M.; Casella, M.; Catto, V.; Pontone, G.; et al. Cardiac mesenchymal stromal cells are a source of adipocytes in arrhythmogenic cardiomyopathy. Eur. Hear. J. 2015, 37, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; On behalf of the ACMG Laboratory Quality Assurance Committee; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Pilato, C.A.; Stadiotti, I.; Maione, A.S.; Saverio, V.; Catto, V.; Tundo, F.; Russo, A.D.; Tondo, C.; Pompilio, G.; Casella, M.; et al. Isolation and Characterization of Cardiac Mesenchymal Stromal Cells from Endomyocardial Bioptic Samples of Arrhythmogenic Cardiomyopathy Patients. J. Vis. Exp. 2018, e57263. [Google Scholar] [CrossRef] [PubMed]
- Casella, M.; Pizzamiglio, F.; Russo, A.D.; Carbucicchio, C.; Al-Mohani, G.; Russo, E.; Notarstefano, P.; Pieroni, M.; D’Amati, G.; Sommariva, E.; et al. Feasibility of Combined Unipolar and Bipolar Voltage Maps to Improve Sensitivity of Endomyocardial Biopsy. Circ. Arrhythmia Electrophysiol. 2015, 8, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Dorn, T.; Kornherr, J.; Parrotta, E.I.; Zawada, D.; Ayetey, H.; Santamaria, G.; Iop, L.; Mastantuono, E.; Sinnecker, D.; Goedel, A.; et al. Interplay of cell–cell contacts and RhoA/ MRTF -A signaling regulates cardiomyocyte identity. EMBO J. 2018, 37, e98133. [Google Scholar] [CrossRef] [PubMed]
- Forkmann, M.; Tomala, J.; Huo, Y.; Mayer, J.; Christoph, M.; Wunderlich, C.; Salmas, J.; Gaspar, T.; Piorkowski, C. Epicardial Ventricular Tachycardia Ablation in a Patient With Brugada ECG Pattern and Mutation of PKP2 and DSP Genes. Circ. Arrhythmia Electrophysiol. 2015, 8, 505–507. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Allegue, C.; Coll, M.; Matés, J.; Campuzano, O.; Iglesias, A.; Sobrino, B.; Brion, M.; Amigo, J.; Carracedo, A.; Brugada, P.; et al. Genetic Analysis of Arrhythmogenic Diseases in the Era of NGS: The Complexity of Clinical Decision-Making in Brugada Syndrome. PLOS ONE 2015, 10, e0133037. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, O.; Fernández-Falgueras, A.; Iglesias, A.; Brugada, R. Brugada Syndrome and PKP2: Evidences and uncertainties. Int. J. Cardiol. 2016, 214, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Tsarouhas, K.; Papalexis, P.; Kafantaris, I.; Tsitsimpikou, C.; Vavetsi, S.; Rentoukas, E. Electrocardiographic findings compatible with Brugada syndrome in a patient with febrile respiratory infection. Hippokratia 2010, 14, 221–223. [Google Scholar] [PubMed]
- Hosseini, S.M.; Kim, R.; Udupa, S.; Costain, G.; Jobling, R.; Liston, E.; Jamal, S.M.; Szybowska, M.; Morel, C.F.; Bowdin, S.; et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death. Circulation 2018, 138, 1195–1205. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Persampieri, S.; Pilato, C.A.; Sommariva, E.; Maione, A.S.; Stadiotti, I.; Ranalletta, A.; Torchio, M.; Dello Russo, A.; Basso, C.; Pompilio, G.; et al. Clinical and Molecular Data Define a Diagnosis of Arrhythmogenic Cardiomyopathy in a Carrier of a Brugada-Syndrome-Associated PKP2 Mutation. Genes 2020, 11, 571. https://doi.org/10.3390/genes11050571
Persampieri S, Pilato CA, Sommariva E, Maione AS, Stadiotti I, Ranalletta A, Torchio M, Dello Russo A, Basso C, Pompilio G, et al. Clinical and Molecular Data Define a Diagnosis of Arrhythmogenic Cardiomyopathy in a Carrier of a Brugada-Syndrome-Associated PKP2 Mutation. Genes. 2020; 11(5):571. https://doi.org/10.3390/genes11050571
Chicago/Turabian StylePersampieri, Simone, Chiara Assunta Pilato, Elena Sommariva, Angela Serena Maione, Ilaria Stadiotti, Antonio Ranalletta, Margherita Torchio, Antonio Dello Russo, Cristina Basso, Giulio Pompilio, and et al. 2020. "Clinical and Molecular Data Define a Diagnosis of Arrhythmogenic Cardiomyopathy in a Carrier of a Brugada-Syndrome-Associated PKP2 Mutation" Genes 11, no. 5: 571. https://doi.org/10.3390/genes11050571
APA StylePersampieri, S., Pilato, C. A., Sommariva, E., Maione, A. S., Stadiotti, I., Ranalletta, A., Torchio, M., Dello Russo, A., Basso, C., Pompilio, G., Tondo, C., & Casella, M. (2020). Clinical and Molecular Data Define a Diagnosis of Arrhythmogenic Cardiomyopathy in a Carrier of a Brugada-Syndrome-Associated PKP2 Mutation. Genes, 11(5), 571. https://doi.org/10.3390/genes11050571