Abstract
The inter-subspecific crossing between indica and japonica subspecies in rice have been utilized to improve the yield potential of temperate rice. In this study, a comparative study of the genomic regions in the eight high-yielding varieties (HYVs) was conducted with those of the four non-HYVs. The Next-Generation Sequencing (NGS) mapping on the Nipponbare reference genome identified a total of 14 common genomic regions of japonica-originated alleles. Interestingly, the HYVs shared japonica-originated genomic regions on nine chromosomes, although they were developed through different breeding programs. A panel of 94 varieties was classified into four varietal groups with 38 single nucleotide polymorphism (SNP) markers from 38 genes residing in the japonica-originated genomic regions and 16 additional trait-specific SNPs. As expected, the japonica-originated genomic regions were only present in the japonica (JAP) and HYV groups, except for Chr4-1 and Chr4-2. The Wx gene, located within Chr6-1, was present in the HYV and JAP variety groups, while the yield-related genes were conserved as indica alleles in HYVs. The japonica-originated genomic regions and alleles shared by HYVs can be employed in molecular breeding programs to further develop the HYVs in temperate rice.
1. Introduction
There are two subspecies in cultivated rice (Oryza sativa), indica and japonica. Indica rice is known to be adaptable to tropical regions, while japonica rice is grown in temperate regions. Therefore, indica and japonica have different characteristics [1]. In general, japonica varieties are known to have relatively low yield potentials, as compared to indica varieties. To improve the yield potential of japonica rice, inter-subspecific crosses between indica and japonica have been conducted by conventional rice breeders [2]. As a result of these efforts, several high-yielding varieties (HYVs) have been developed from indica-japonica crosses. One of the greatest historical successes of indica–japonica crosses was the development of Tongil, an HYV, in Korea. Tongil showed a 30% higher yield than those of the conventional japonica varieties. By growing Tongil rice, self-sufficiency in staple food in Korea was possible in 1977 [3]. There were important trade-offs in growing Tongil, such as cold intolerance, pathogen susceptibility, and low eating quality, which were inherited from indica parents. A series of ‘Tongil-type’ HYVs have been developed from indica–japonica crosses to overcome the vulnerable points of Tongil from the late 1970s [2].
HYVs have been developed in Japan using indica and japonica varieties since the 1980s. Takanari is a Japanese semi-dwarf HYV, developed from the crosses between Milyang 42 and Milyang 25. Takanari shared the ancestry of Tongil [4]. To date, it recorded the highest grain yields for both yield trials (>10 t/ha as brown rice) and individual trials (11.7 t/ha as brown rice) in Japan [5]. Minghui 63, which was derived from the cross between IR 30 and Gui 630, is the male parent of the elite hybrid rice Shanyu 63 from China. IR 30 is a semi-dwarf variety developed in IRRI (International Rice Research Institute) and is a restorer line for WA-CMS A-lines, which have a good plant type, a high resistance to blast, a bacterial blight, and brown planthoppers. Gui 630 is a rice germplasm from Guyana that has a high grain weight, desirable grain quality, and high yield potential [6]. Gui 630 is known as an indica restorer variety [7]. Minghui 63 was classified into the indica II subpopulation, together with Milyang 23 and some other indica-japonica HYVs, by genome sequence analysis [8].
Nipponbare is the japonica reference genome of rice and was first sequenced at the high-quality whole-genome level through all the crops [9]. In addition, the whole genome sequences of indica rice varieties were reported [10,11,12,13,14]. The genomic difference between indica and japonica at the sequence level has been extensively studied [15]. At least 384,431 single nucleotide polymorphisms (SNPs) and 24,557 insertion/deletion mutations (InDels) were reported between Nipponbare and 93-11 [16]. With the advent of Next-Generation Sequencing (NGS) technology, numerous genomes of diverse rice germplasm collections have become available. For instance, 3000 rice genomes were sequenced and deployed in genetic and genomic rice studies [17,18,19,20]. Recently, more than one hundred high-yielding loci, associated with green revolution phenotypes and derived from the two ancestral indica varieties, were identified with the help of pedigree analysis, whole-genome sequencing, and genome editing [21]. Furthermore, most of the quantitative trait loci (QTLs) and genes for high-yielding potential in HYVs originated from indica parents in previous studies using HYVs derived from indica–japonica crosses [3,4,5,22,23]. No report exists on the characterization of japonica genomic regions in HYVs derived from indica–japonica crosses yet.
Previously, we sequenced the whole genomes of Tongil and its parental varieties to analyze the genome composition and genetic factors of Tongil. As a result, the Tongil genome was found to be derived mostly from the indica genome, with a small portion of japonica genome introgressions [3]. This study was carried out to comparatively analyze the genome structure of eight HYVs and to identify the japonica-originated genomic regions that are shared in HYVs, which will be helpful in understanding the role of japonica genome in Tongil and other HYVs that are developed in temperate regions for the further development of promising HYVs.
2. Materials and Methods
2.1. Plant DNA Materials
The eight HYVs, including Cheongcheongbyeo, Dasanbyeo, Hanareumbyeo, Milyang 23, Minghui 63, Nampungbyeo, Takanari, and Tongil, and four non-HYVs, including Nipponbare, Yukara, IR 8, and TN1 were used for the whole-genome sequence analysis. Cheongcheongbyeo, Dasanbyeo, Hanareumbyeo, Milyang 23, and Nampungbyeo are Tongil-type HYVs that were developed in Korea. Takanari and Minghui 63 are HYVs from Japan and China, respectively. The pedigree of each of the eight HYVs can be found in Figure S1. A total of 94 rice varieties were used for SNP marker validation (Table S1).
2.2. Whole Genome Sequencing and DNA Variation
Tongil and its three parental varieties (Yukara, IR 8, and TN1) were sequenced in a previous study [3]. The other eight varieties were sequenced in this study using the Illumina Hiseq 1000 and NextSeq 500 platform (Illumina, San Diego, CA, USA) (Table 1). Whole genome sequencing, including the construction of shotgun DNA libraries, was performed according to the methods recommended by the manufacturer. The Illumina whole-genome shotgun paired-end DNA sequencing data were filtered to obtain high-quality sequence data. Raw sequence reads were subjected to quality trimming using FastQC v0.11.3 (http://www.bioinformatics.babraham.ac.uk/ projects/fastqc/), and the reads with a Phred quality (Q) score <20 were discarded. Adapter trimming was conducted by using Trimmomatic version 0.36 (http://www.usadellab.org/cms/?page=trimmomatic).
The clean reads were mapped on the japonica reference Nipponbare genome (Os-Nipponbare-Reference-IRGSP-1.0 [24]) using the Burrows–Wheeler Aligner (BWA) program version 0.7.15 [25]. The alignment results were merged and converted into binary alignment map (BAM) files [26]. The BAM files were used to calculate the sequencing depth and to identify SNPs using the GATK program version 3.4 with default parameters [27].
All the raw sequence data obtained in this study are available in the NCBI Short Read Archive (SRA) database under the following BioProject accession numbers: Nipponbare [PRJNA264254], Milyang 23 [PRJNA264250], Dasan (or Dasanbyeo) [PRJNA222717], Cheongcheong (or Cheongcheongbyeo) [PRJNA616202], Nampung (or Nampungbyeo) [PRJNA616219], Hanareum (or Hanareumbyeo) [PRJNA616209], Takanari [PRJNA616222], and Minghui 63 [PRJNA616216]. The raw sequence data of Tongil, Yukara, IR 8, and TN1 are available in a previous study [3].
2.3. SNP Allele Calling
Genotype calling was performed in order to identify SNPs that originated from the indica and japonica genomes. There were two types of values calculated in this study: variety value and reference value. The variety value calculated whether it was of japonica-type parental allele (Yukara allele) of Tongil or a Tongil-like variety or not. The variety value of SNP was calculated as the sum of the following values: ‘1’ (IR 8 allele); ‘2’ (TN1 allele); ‘4’ (Yukara allele); and ‘0’ (all others). If the SNP is the same as the Nipponbare reference, the value ‘1’ was given to the SNP; otherwise, the value ‘0’ was given. The SNPs showing variety value ‘4’ and reference value ‘1’ were called japonica-type SNPs. Then, the total number of japonica-type SNPs in each 100 kb block, which is the approximate chromosomal distance for the linkage disequilibrium (LD) decay rate in rice [28], in each chromosome was counted to identify the introgressed regions of japonica.
2.4. SNP Marker Development for the Fluidigm Platform
A total of 39 representative genes were selected from the selected regions. The representative genes were well-annotated in the public gene/QTL databases, as of 6 January 2017 (RAP and UniProt). Then, only the genes containing non-synonymous SNPs in the predicted exon and UTR regions between HYVs and non-HYVs were selected. We assumed that these genes might be a part of the candidate genes that are associated with japonica-originated traits. Out of the many SNPs in the genes, only one SNP with a substitution polymorphism between indica (IR 8 and TN1) and japonica (Nipponbare and Yukara) per representative gene was selected for the genomic validation. The SNP markers for the Fluidigm platform (Fluidigm Corporation, San Francisco, CA, USA) were designed using the method by Seo et al. [29]. To design Fluidigm SNP genotyping assays, 60–150 bp sequences, flanking the selected SNPs on either side, were aligned by BLAST. Finally, the selected SNPs and flanking sequences were uploaded on the D3 Assay Design website [30]. After confirming the results, the designed assays were ordered. One Fluidigm SNP assay contained the Allele-Specific Primer 1 (ASP1), ASP2, Locus-Specific Primer (LSP), and Specific Target Amplification (STA) primer.
2.5. DNA Extraction and Fluidigm Genotyping
Young leaves of each plant from all materials used in this study were collected for DNA extraction at the tillering stage. Genomic DNA was extracted using the modified cetyltrimethylammonium bromide (CTAB) method, as described by Murray and Thompson [31]. The concentration and purity of DNA samples were measured with a NanoDrop 1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). DNA samples, showing absorbance ratios above 1.8 at 260/280 nm, were diluted to 50 ng/μL and used for genotyping.
Genotyping was performed using the BioMark™ HD system (Fluidigm) and 96.96 Dynamic Array IFCs (Fluidigm), according to the manufacturer’s protocol in National Instrumentation Center for Environmental Management (NICEM), Seoul National University (Pyeongchang, Korea). Specific Target Amplification (STA) was performed prior to SNP genotyping analysis. PCR was performed in a 5 μL reaction containing 50 ng of the DNA sample, according to the manufacturer’s protocol. For genotyping, SNPtype assays were performed using STA products, according to the manufacturer’s protocol. The genotyping result was acquired using Fluidigm SNP Genotyping Analysis software. All the genotype-calling results were manually checked and any obvious errors in the homozygous or heterozygous clusters were curated.
2.6. Data Analysis and QTL Comparison
We analyzed basic marker statistics, such as major allele frequency (MAF), heterozygosity, and polymorphism information content (PIC) of SNP markers using PowerMarker V3.25 [32]. PowerMarker V3.25 was used to calculate the genetic distance, based on CS Chord [33] and the constructed un-weighted pair group methods with the arithmetic mean algorithm (UPGMA) dendrogram, which was visualized in Molecular Evolutionary Genetics Analysis version 7.0; MEGA7 [34].
We extracted QTL information from the Q-TARO database at 25th August 2017. The physical position in Q-TARO is based on IRGSP build 4 of the Nipponbare genome, while IRGSP 1.0 of the Nipponbare genome was used in this study. Thus, the physical position of the start and end of each QTLs in Q-TARO were converted into the physical positions of IRGSP 1.0. Then, the QTLs overlapped on common japonica-originated regions of eight HYVs were selected. After checking if all the selected QTLs were unique or redundant, the redundant QTLs were discarded and only the unique QTLs were left remained. After the filtering step for the QTLs, they were classified into seven categories. The category classification was conducted by checking the characters and the trait names manually.
3. Results
3.1. Whole Genome Sequencing and SNP Calling
To analyze the genomic composition of the HYVs derived from indica–japonica crosses, the whole genomes of HYVs and four varieties were sequenced on the Illumina platform. A large number of short reads were mapped onto the reference Nipponbare genome and were then assembled into a consensus sequence. The number of sequence yields and the number of reads and mapping depths were varied. For example, a total of 66,464,246 reads of the Cheongcheongbyeo genome, corresponding to 9,991,040,272 bp (10 Gb), were generated, representing a 21-fold mapping depth. Nipponbare showed the largest number of sequence yields and number of reads and mapping depths (Table 1).

Table 1.
Basic sequencing statistics of the varieties used in this study.
More than one million SNPs were detected in each of the eight HYVs and two indica varieties against the Nipponbare (japonica) genome. More than 90% of the SNPs were detected in the intergenic region. The smallest number of SNPs were in the 5′ untranslated region (UTR). Two indica varieties, IR 8 and TN1, represented a relatively large number of SNPs, as compared to seven HYVs except for Minghui 63. Among the seven HYVs, Milyang 23 showed the smallest number of SNPs (Table 2).
Table 2.
Number and location of the SNPs in the varieties against Nipponbare pseudomolecule.
3.2. Evaluation of Japonica-Type SNP Value
To identify the SNPs that originated from the indica and japonica genomes, the SNP value was evaluated. The SNPs showing a variety value of 4 and a reference value of 1 were japonica-originated SNPs. The total number of japonica-originated SNPs in each 100 kb block of each chromosome were counted in order to identify introgressed regions from japonica. Previously, we discriminated the Tongil genome into segments that originated from indica and japonica using the sliding window method [3]. In this study, we allowed for two exception from the HYV in order to include Takanari, Cheongcheong, Nampung and Minghui 63. Accordingly, a total of 14 japonica-originated genomic regions, which were shared by at least six HYVs on japonica-originated segment of Tongil, were detected. The common japonica-originated genomic regions were distributed on nine chromosomes, not including chromosomes 8, 10, and 12. There were three japonica-type regions on chromosome 2 and two regions on chromosomes 1, 4, and 7. Furthermore, the regions were clustered or closely located on each chromosome. The size of the regions was varied from 0.1 Mb for Chr7-1 and Chr11-1 to 2 Mb for Chr1-2. Out of the 14 regions, seven were common in eight HYVs (Figure 1, Table 3).
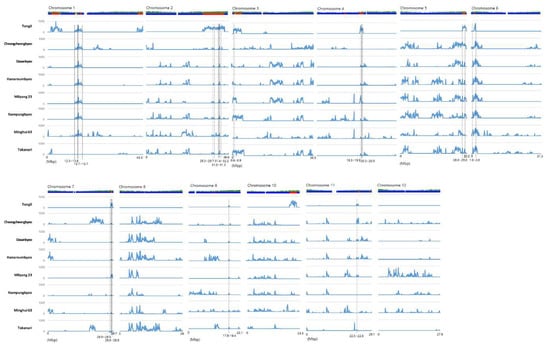
Figure 1.
Location of japonica-type SNPs on 12 chromosomes of HYVs and their co-location with the japonica block of the Tongil genome, shown as a bar above the graphs. The blue and red block of the Tongil genome represents the indica and japonica blocks, respectively. The blue peak on each graph indicates the number of japonica-type SNPs. The position and range for the co-location of japonica-type blocks are denoted by vertical black dotted lines.
Table 3.
Japonica-type SNP frequency (%) of Tongil and the other HYVs at common japonica-type regions among the eight HYVs.
3.3. QTL Comparison and Representative Gene Selection in Common japonica-Originated Genomic Regions
To elucidate the function of common japonica-type regions in HYVs, we first investigated the reported QTLs in the Q-TARO database [35]. A total of 101 selected QTLs for seven categories were co-located, with 14 common japonica-type regions on nine chromosomes. Category classification was carried out by checking the character and trait names of each QTL manually. For instance, the yield-related trait category contained various characters which could affect yield potential, such as source activity- and sink-related morphological traits, and sterility. Only three regions on chromosome 2 were co-located with QTLs for all the seven trait categories. For eating quality, abiotic stress, and the yield-related category, 80 QTLs were identified. The largest number (10) of co-located QTLs was detected in the Chr6-1 region for eating quality. All the regions were co-located with the QTLs for abiotic stress tolerance (Table 4). This information of co-located QTLs, with common japonica-type regions, suggests that common genomic regions in HYVs might be mainly associated with quality, yield, and abiotic stress tolerance.
Table 4.
Classification of the reported QTLs co-location with common japonica-type regions in eight HYVs.
Furthermore, we selected 39 genes containing non-synonymous SNPs, which could affect the molecular function of genes, and are clearly annotated in the databases of 12 common japonica chromosomal introgressions. There was no target gene that satisfied the above-mentioned condition in Chr4-2 and Chr7-1. The largest number (13) of selected genes was located on Chr1-2, which is the largest region, spanning 2 Mb. Only one gene was selected from Chr2-2, Chr7-2, and Chr11-1. The size of these three blocks was 0.1–0.2 Mb. The genes annotated from the major criteria of interest were Os01g0348900, Os06g0130000, Os06g0130100 (stress tolerance), Os06g0130400 (eating quality), and Os01g0367100 (yield potential) (Table 5).
Table 5.
Selected 39 genes in the common japonica-type regions.
3.4. SNP Marker Development and Genotyping Using Fluidigm Platform
A total of 39 SNP markers were designed in the 39 selected genes from common japonica-originated genomic regions, by one marker per one gene. The SNPs for the marker were selected from among the non-synonymous SNPs. Five SNP markers, out of the 39 SNP markers, were designed in 3′ or 5′ UTR. In addition, 14 agronomic traits, related to SNP markers in indica-japonica SNP set 2 [29] and four previously developed yield related SNP markers [36], were also used for the genotyping of 94 diverse germplasms. A total of 57 SNP markers were genotyped for 94 germplasms using the Fluidigm system, and consequently, 54 SNPs showed polymorphism and a clear genotype, except one monomorphic SNP marker designed in Os01g0348900 on the Chr1-2 block and two SNP markers which showed low base call quality, SaF-CT and SLG7-GC, in indica-japonica SNP set 2. Therefore, we conducted a further analysis using a total of 54 polymorphic SNP markers (Figure 2, Table S2).
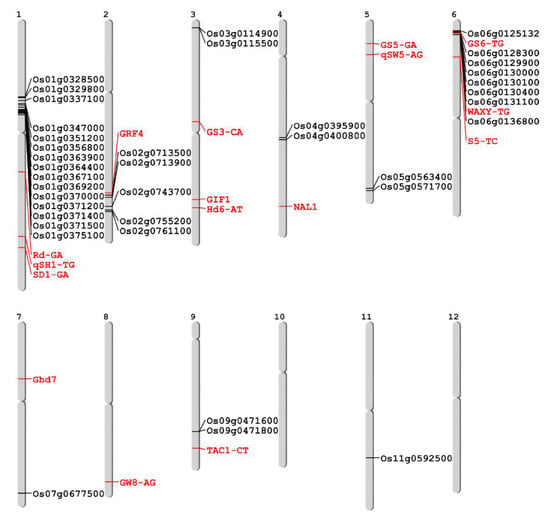
Figure 2.
Genomic location of 54 polymorphic SNP markers used in this study. The markers represented by black and red indicate those newly developed on common japonica regions and those previously developed for agronomic trait related genes, respectively.
The results of genotyping showed a dividing pattern for 94 varieties. A phylogenetic analysis of 94 varieties was carried out using 54 polymorphic SNP markers. There were four groups, including IND1, IND2, HYV, and JAP, in the phylogenetic tree (Figure 3). All the sequenced HYVs, except Takanari, which possesses indica allele of Chr1-1 and Chr1-2, were clustered in the HYV-group with seven Tongil-type and five indica varieties. These varieties were developed by inter-subspecific crosses or by inter-varietal crosses including the parents of Tongil-type varieties. For example, Keunseombyeo was derived from a cross between Dasanbyeo and Namyeongbyeo [37]. Taebaekbyeo was used for the development of Hanareumbyeo and Dasanbyeo (Figure S1, F–G). On the other hand, among the five indica varieties, four indica (IR24, IRBB23, IRBB61, and IRBB66) were developed by the International Rice Research Institute (IRRI). IRBB23, IRBB61, and IRBB66 are near-isogenic line series of IR24 for the improvement of the resistance against bacterial leaf blight [38]. IR24 is a good eating quality and high-yielding variety developed from the inter-subspecific crosses using one tropical japonica (CP-SLO) and two indica (SIGADIS and IR8) [39]. Furthermore, IR24 was included in the breeding program of six HYVs, which were used for resequencing in this study. (Figure S1, C–H). This implies that the specific genomic regions were conserved in the HYV group by the selections during the conventional breeding programs for HYV development.
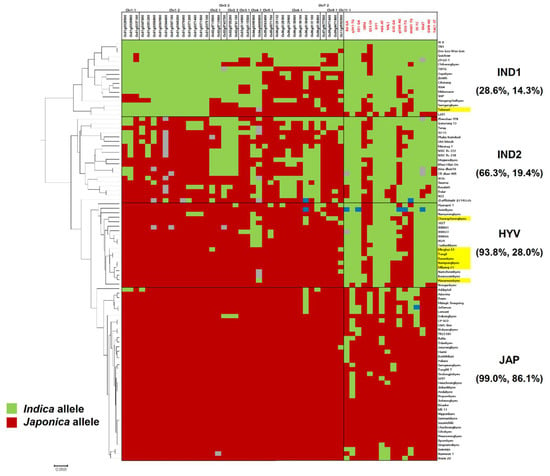
Figure 3.
Phylogenetic tree of 94 germplasms based on 54 SNP markers and a genotype heat map. A 38 SNP marker set (left) and 16 SNP marker set (right) were developed on common japonica regions and agronomic trait related genes, respectively. The color of the marker ID is the same as in Figure 2. The varieties highlighted with yellow are eight resequenced HYVs. Homozygous alleles, which are identical to Nipponbare, were represented as red, and this is different from Nipponbare, represented as green and heterozygous alleles as blue. Grey indicates a missing genotype. The percentage values in parenthesis under each subgroup represent the percentage for homozygous indica allele in the 38 SNP marker set and 16 SNP marker set.
A total of 38 SNP markers were developed in the common Tongil-like japonica-like regions and discriminated IND1, IND2, and HYV by the frequency of japonica-alleles. The HYV group showed 93.8% for japonica-alleles frequency, which is similar to that of JAP. A total of 16 SNP markers were related to yield and some agronomic traits could distinguish JAP from the other three groups. For the 16 SNP set, the JAP group represented 86.1% of japonica allele, while the other three groups showed a lower japonica allele frequency, which was less than 30%. The HYV group contained indica alleles and were informed by the makers that were linked to the genes associated with plant architecture (SD1-GA, NAL1, and TAC-CT), yield potential (GIF1, Hd6-AT, Ghd7, and GW8-AG), and subspecies differentiation (Rd-GA, qSH1-TG, and S5-TC). They contained more than 50% japonica alleles using the markers linked to the genes for grain shape and quality (GRF4, GS3-CA, qSW5-AG, GS6-GT, and WAXY-TG) of HYV-type. Practically, the markers designed in the japonica-originated genomic regions and the yield-related markers from indica varieties can differentiate HYV from indica and japonica varieties. In fact, a high proportion of japonica alleles on Chr1-1, Chr1-2, and Chr3-1 were found in IND2, which consist of three aus varieties, one wild rice relative accession, and one weedy rice variety.
4. Discussion
All eight HYVs used in this study were clustered into the indica group, based on indica–japonica SNP sets reported in a previous study [29]. Interestingly, some japonica-type SNPs were detected within the genomes of HYVs after resequencing analysis. Furthermore, collocations of 14 japonica-originated genomic regions commonly present in Tongil-like HYVs, inherited from indica–japonica crosses, were investigated. This suggests that these japonica-type genomic segments were commonly and repeatedly selected during independent breeding programs in the temperate rice cultivation area. To investigate the role of these japonica segments in HYVs, a comparative study of the reported QTLs and representative gene selection were conducted. Eating quality, stress tolerance, and yield related traits might be main drivers for the selection of the rice HYV breeding program.
A total of 54 SNP markers, including 38 SNP markers developed from 38 selected genes for 14 common japonica-type regions and 16 trait-specific SNP markers, were used for the genotyping of 94 diverse varieties across indica and japonica. For the 38 SNP marker set, 16 SNP markers were located on chromosome 1, which was not identified as a japonica genomic segment in Takanari. Consequently, Takanari was clustered into the IND1 subgroup, although it showed a similar genotypic pattern to the other HYVs. The Chr1-1 and Chr1-2 regions were co-located with some abiotic tolerance and yield related QTLs. Furthermore, there were several selected genes that conferred abiotic stress tolerance and yield potential in the blocks. For instance, Os01g0337100 (OsTPS1) reported an association with abiotic stress response and tolerance by knock-out and overexpression [40,41]. Os01g0367100 (PHD1) was shown to be a gene that was involved in galactolipid biosynthesis and affected photosynthetic efficiency [42]. Recently, the effect of the haplotype of PHD1 on grain yield was also reported using the 3K rice genome panel [18]. Takanari could not have acquired this japonica genomic segment from a different natural environment and/or breeder’s selection.
In addition, 18 varieties out of the 19 HYV-types, showed japonica-type Wxb allele on the SNP marker WAXY-TG, which was designed on the splicing site in the intron of Wx gene in the japonica-type region. The Wx gene only contains synonymous SNPs, although it is located within Chr6-1; thus, it was not selected in our study. We previously developed a functional SNP marker for the Wx gene [29]. The genomic region containing the Wx gene is a hotspot for grain quality [43] and has been selected during and after the domestication of rice [44]. The other genomic research using two Tongil-type varieties also showed a japonica-type SNP pattern on the common japonica-type region on chromosome 6 [45]. Tongil-type varieties showed medium amylose contents, approximately 19–20%, which is similar to that of non-waxy Korean japonica varieties [46,47]. Further, Os06g0130400, one of the selected genes in Chr6-1, was also reported as the gene controlling starch grain size in endosperm [48]. Os06g0130000 and Os06g0130100 were reported for resistance to rice blast and bacterial blight, and tolerance to drought and salt stress, respectively [49,50]. Therefore, Chr6-1, including the Wx gene, might be mainly selected for eating quality and latent stress tolerance.
When HYVs were developed by inter-subspecific hybridization, the breeders aimed at not only transferring some of the desirable characteristics, like resistance to lodging, blast, and yield, but also at retaining the ecological adaptability and eating quality of japonicas [51]. The japonica chromosomal introgression regions identified in this study were regarded as putative temperate region adaptable and improved the eating quality of indica. For this reason, the varieties developed by indica–japonica crosses could also be considered as ‘temperate indica’. Recently, new elite rice varieties showing high yield potential and high grain quality were developed by the precise pyramiding of major genes controlling yield and grain quality traits [52]. Furthermore, there was an attempt to develop cold tolerant indica using an inter-subspecific cross and marker-assisted selection (MAS) [53]. In other words, breeding indica varieties, which are adaptable to the temperate region with high yield potential and good eating quality, can be efficiently achieved through inter-subspecific crosses and marker-assisted selection using the SNP markers developed in this study. Nevertheless, to dissect the exact contribution of japonica-type regions in HYVs, a comprehensive genetic and physiological analysis, by applying the molecular markers developed in japonica-type regions to the segregation populations derived from cross between HYVs and indica, is necessary. In addition, the functional studies of genes in the regions, as well as the selected ones in this study, are also required.
5. Conclusions
Consumer preference in grain shape and quality during conventional breeding procedures, without sacrificing the high-yield potential of indica, were revisited by the genomic analysis of HYVs. The 14 japonica-originated genomic regions and alleles identified in this study shared by HYVs could be applied in further development of more HYVs through inter-subspecific rice breeding in temperate rice.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4425/11/5/562/s1, Figure S1: Pedigrees of eight HYVs used in this study. (A) Tongil, (B) Minghui 63, (C) Cheongcheongbyeo, (D) Milyang 23, (E) Nampungbyeo, (F) Hanareumbyeo, (G) Dasanbyeo, (H) Takanari. Table S1: The list of 94 diverse rice germplasms used in this study. The order of ger1mplasm is identical with order in Figure 3. The varieties highlighted by yellow are eight HYVs used in this study. Table S2: The list of 54 Fluidigm SNP markers used in this study. MAF, Heterozygosity and PIC were calculated in 94 rice varieties.
Author Contributions
Conceptualization, J.H.C. and H.-J.K.; methodology, J.S. and J.H.C.; software, J.S.; validation, B.K. and J.H.C.; formal analysis, J.S. and S.-M.L.; investigation, J.S., S.-M.L., J.-H.H., N.-H.S., and Y.K.L., resources, J.H.C and H.-J.K.; data curation, J.S. and B.K.; writing—original draft preparation, J.S.; writing—review and editing, J.H.C. and H.-J.K.; visualization, J.S., S.-M.L., and J.H.C.; supervision, H.-J.K.; project administration, J.H.C.; funding acquisition, H.-J.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by a grant from the Next-Generation BioGreen 21 Program (No. PJ013165) of the Rural Development Administration, Korea.
Acknowledgments
We appreciate to members of Crop Molecular Breeding Lab., in Seoul National University for their kind advice and assistance for this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Oka, H.I. Origin of Cultivated Rice; Japan Scientific Societies Press: Tokyo, Japan, 1988. [Google Scholar]
- Chung, G.S.; Heu, M.H. Improvement of Tongil-Type Rice Cultivars from Indica/Japonica Hybridization in Korea. In Rice; Bajaj, Y.P.S., Ed.; Springer: Berlin/Heidelberg, Germany, 1991; pp. 105–112. [Google Scholar] [CrossRef]
- Kim, B.; Kim, D.-G.; Lee, G.; Seo, J.; Choi, I.-Y.; Choi, B.-S.; Yang, T.-J.; Kim, K.; Lee, J.; Chin, J.; et al. Defining the genome structure of ‘Tongil’ rice, an important cultivar in the Korean “Green Revolution”. Rice 2014, 7, 22. [Google Scholar] [CrossRef]
- Takai, T.; Arai-Sanoh, Y.; Iwasawa, N.; Hayashi, T.; Yoshinaga, S.; Kondo, M. Comparative Mapping Suggests Repeated Selection of the Same Quantitative Trait Locus for High Leaf Photosynthesis Rate in Rice High-Yield Breeding Programs. Crop Sci. 2012, 52, 2649–2658. [Google Scholar] [CrossRef]
- Takai, T.; Ikka, T.; Kondo, K.; Nonoue, Y.; Ono, N.; Arai-Sanoh, Y.; Yoshinaga, S.; Nakano, H.; Yano, M.; Kondo, M.; et al. Genetic mechanisms underlying yield potential in the rice high-yielding cultivar Takanari, based on reciprocal chromosome segment substitution lines. BMC Plant Biol. 2014, 14, 295. [Google Scholar] [CrossRef]
- Xie, F.; Zhang, J. Shanyou 63: An elite mega rice hybrid in China. Rice 2018, 11, 17. [Google Scholar] [CrossRef]
- Zhu, L.; Lu, C.; Li, P.; Shen, L.; Xu, Y.; He, P.; Chen, Y. Using doubled haploid populations of rice for quantitative trait locus mapping. In Rice Genetics III. Proceedings of the Third International Rice Genetics Symposium, Manila, Philippines, 16–20 October 1995; Khush, G.S., Ed.; International Rice Research Institute: Los Baños, Philippines, 1996; pp. 631–636. [Google Scholar]
- Xie, W.; Wang, G.; Yuan, M.; Yao, W.; Lyu, K.; Zhao, H.; Yang, M.; Li, P.; Zhang, X.; Yuan, J.; et al. Breeding signatures of rice improvement revealed by a genomic variation map from a large germplasm collection. Proc. Natl. Acad. Sci. USA 2015, 112, E5411–E5419. [Google Scholar] [CrossRef]
- Sasaki, T. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.-S.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A Draft Sequence of the Rice Genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef]
- Schatz, M.C.; Maron, L.G.; Stein, J.C.; Wences, A.H.; Gurtowski, J.; Biggers, E.; Lee, H.; Kramer, M.; Antoniou, E.; Ghiban, E.; et al. Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica. Genome Biol. 2014, 15, 1–16. [Google Scholar] [CrossRef]
- Sakai, H.; Kanamori, H.; Arai-Kichise, Y.; Shibata-Hatta, M.; Ebana, K.; Oono, Y.; Kurita, K.; Fujisawa, H.; Katagiri, S.; Mukai, Y.; et al. Construction of Pseudomolecule Sequences of the aus Rice Cultivar Kasalath for Comparative Genomics of Asian Cultivated Rice. DNA Res. 2014. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, L.-L.; Xing, F.; Kudrna, D.A.; Yao, W.; Copetti, D.; Mu, T.; Li, W.; Song, J.-M.; Xie, W.; et al. Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63. Proc. Natl. Acad. Sci. USA 2016, 113, E5163–E5171. [Google Scholar] [CrossRef]
- Du, H.; Yu, Y.; Ma, Y.; Gao, Q.; Cao, Y.; Chen, Z.; Ma, B.; Qi, M.; Li, Y.; Zhao, X.; et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat. Commun. 2017, 8, 15324. [Google Scholar] [CrossRef]
- Subbaiyan, G.K.; Waters, D.L.; Katiyar, S.K.; Sadananda, A.R.; Vaddadi, S.; Henry, R.J. Genome-wide DNA polymorphisms in elite indica rice inbreds discovered by whole-genome sequencing. Plant Biotechnol. J. 2012, 10, 623–634. [Google Scholar] [CrossRef]
- Feltus, F.A.; Wan, J.; Schulze, S.R.; Estill, J.C.; Jiang, N.; Paterson, A.H. An SNP resource for rice genetics and breeding based on subspecies indica and japonica genome alignments. Genome Res. 2004, 14, 1812–1819. [Google Scholar] [CrossRef]
- Sun, C.; Hu, Z.; Zheng, T.; Lu, K.; Zhao, Y.; Wang, W.; Shi, J.; Wang, C.; Lu, J.; Zhang, D.; et al. RPAN: Rice pan-genome browser for approximately 3000 rice genomes. Nucleic Acids Res. 2017, 45, 597–605. [Google Scholar] [CrossRef]
- Abbai, R.; Singh, V.K.; Nachimuthu, V.V.; Sinha, P.; Selvaraj, R.; Vipparla, A.K.; Singh, A.K.; Singh, U.M.; Varshney, R.K.; Kumar, A. Haplotype analysis of key genes governing grain yield and quality traits across 3K RG panel reveals scope for the development of tailor-made rice with enhanced genetic gains. Plant Biotechnol. J. 2019, 17, 1612–1622. [Google Scholar] [CrossRef]
- Carpentier, M.C.; Manfroi, E.; Wei, F.J.; Wu, H.P.; Lasserre, E.; Llauro, C.; Debladis, E.; Akakpo, R.; Hsing, Y.I.; Panaud, O. Retrotranspositional landscape of Asian rice revealed by 3000 genomes. Nat. Commun. 2019, 10, 24. [Google Scholar] [CrossRef]
- Fuentes, R.R.; Chebotarov, D.; Duitama, J.; Smith, S.; De la Hoz, J.F.; Mohiyuddin, M.; Wing, R.A.; McNally, K.L.; Tatarinova, T.; Grigoriev, A.; et al. Structural variants in 3000 rice genomes. Genome Res. 2019, 29, 870–880. [Google Scholar] [CrossRef]
- Huang, J.; Li, J.; Zhou, J.; Wang, L.; Yang, S.; Hurst, L.D.; Li, W.H.; Tian, D. Identifying a large number of high-yield genes in rice by pedigree analysis, whole-genome sequencing, and CRISPR-Cas9 gene knockout. Proc. Natl. Acad. Sci. USA 2018, 115, E7559–E7567. [Google Scholar] [CrossRef]
- Adachi, S.; Yamamoto, T.; Nakae, T.; Yamashita, M.; Uchida, M.; Karimata, R.; Ichihara, N.; Soda, K.; Ochiai, T.; Ao, R.; et al. Genetic architecture of leaf photosynthesis in rice revealed by different types of reciprocal mapping populations. J. Exp. Bot. 2019, 70, 5131–5144. [Google Scholar] [CrossRef]
- Takai, T.; Adachi, S.; Taguchi-Shiobara, F.; Sanoh-Arai, Y.; Iwasawa, N.; Yoshinaga, S.; Hirose, S.; Taniguchi, Y.; Yamanouchi, U.; Wu, J.; et al. A natural variant of NAL1, selected in high-yield rice breeding programs, pleiotropically increases photosynthesis rate. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Barnett, D.W.; Garrison, E.K.; Quinlan, A.R.; Stromberg, M.P.; Marth, G.T. BamTools: A C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 2011, 27, 1691–1692. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef]
- Seo, J.; Lee, G.; Jin, Z.; Kim, B.; Chin, J.H.; Koh, H.-J. Development and application of indica–japonica SNP assays using the Fluidigm platform for rice genetic analysis and molecular breeding. Mol. Breed. 2020, 40, 39. [Google Scholar] [CrossRef]
- D3 Assay Design-Fluidigm. Available online: https://d3.fluidigm.com (accessed on 25 September 2019).
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef]
- Cavalli-Sforza, L.L.; Edwards, A.W.F. Phylogenetic analysis. Models and estimation procedures. Am. J. Hum. Genet. 1967, 19, 233–257. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Yonemaru, J.-i.; Yamamoto, T.; Fukuoka, S.; Uga, Y.; Hori, K.; Yano, M. Q-TARO: QTL Annotation Rice Online Database. Rice 2010, 3, 194–203. [Google Scholar] [CrossRef]
- Seo, J.; Lee, S.-M.; Han, J.-H.; Shin, N.-H.; Koh, H.-J.; Chin, J.H. Identification of Yield and Yield-Related Quantitative Trait Loci for the Field High Temperature Condition in Backcross Populations of Rice (Oryza sativa L.). Plant Breed. Biotechnol. 2019, 7, 415–426. [Google Scholar] [CrossRef]
- Ha, U.-G.; Song, Y.-C.; Yeo, U.-S.; Cho, J.-H.; Hwang, H.-G.; Kim, Y.-D.; Cho, Y.-H.; Yang, S.-J.; Lee, J.-H.; Oh, B.-G.; et al. A New High Yielding Rice Variety with Multi-Disease Resistance, ‘Keunseom’. Korean J. Breed. Sci. 2011, 43, 576–580. [Google Scholar]
- Huang, N.; Angeles, E.R.; Domingo, J.; Magpantay, G.; Singh, S.; Zhang, G.; Kumaravadivel, N.; Bennett, J.; Khush, G.S. Pyramiding of bacterial blight resistance genes in rice: Marker-assisted selection using RFLP and PCR. Theor. Appl. Genet. 1997, 95, 313–320. [Google Scholar] [CrossRef]
- Khush, G.; Virk, P. IR Varieties and Their Impact, 1st ed.; International Rice Research Institute: Los Baños, Philippines, 2005; pp. 46–48. [Google Scholar]
- Kim, S.-J.; Jeong, D.-H.; An, G.; Kim, S.-R. Characterization of a drought-responsive gene, OsTPS1, identified by the T-DNA Gene-Trap system in rice. J. Plant Biol. 2005, 48, 371–379. [Google Scholar] [CrossRef]
- Li, H.W.; Zang, B.S.; Deng, X.W.; Wang, X.P. Overexpression of the trehalose-6-phosphate synthase gene OsTPS1 enhances abiotic stress tolerance in rice. Planta 2011, 234, 1007–1018. [Google Scholar] [CrossRef]
- Li, C.; Wang, Y.; Liu, L.; Hu, Y.; Zhang, F.; Mergen, S.; Wang, G.; Schlappi, M.R.; Chu, C. A rice plastidial nucleotide sugar epimerase is involved in galactolipid biosynthesis and improves photosynthetic efficiency. PLoS Genet. 2011, 7, e1002196. [Google Scholar] [CrossRef]
- Sreenivasulu, N.; Butardo, V.M., Jr.; Misra, G.; Cuevas, R.P.; Anacleto, R.; Kavi Kishor, P.B. Designing climate-resilient rice with ideal grain quality suited for high-temperature stress. J. Exp. Bot. 2015, 66, 1737–1748. [Google Scholar] [CrossRef]
- Olsen, K.M.; Caicedo, A.L.; Polato, N.; McClung, A.; McCouch, S.; Purugganan, M.D. Selection Under Domestication: Evidence for a Sweep in the Rice Waxy Genomic Region. Genetics 2006, 173, 975–983. [Google Scholar] [CrossRef]
- Ji, H.; Ahn, E.; Seo, B.; Kang, H.; Choi, I.; Kim, K. Genome-wide detection of SNPs between two Korean Tongil-type rice varieties. Korean J. Breed. Sci. 2016, 48, 460–469. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Yang, C.-I.; Choi, Y.-H.; Won, Y.-J.; Lee, Y.-T. Changes of Seed Viability and Physico-Chemical Properties of Milled Rice with Different Ecotypes and Storage Duration. Korean J. Crop Sci. 2007, 52, 375–379. [Google Scholar]
- Kwak, J.; Yoon, M.R.; Lee, J.S.; Lee, J.H.; Ko, S.; Tai, T.H.; Won, Y.J. Morphological and starch characteristics of the Japonica rice mutant variety Seolgaeng for dry-milled flour. Food Sci. Biotechnol. 2017, 26, 43–48. [Google Scholar] [CrossRef]
- Matsushima, R.; Maekawa, M.; Kusano, M.; Tomita, K.; Kondo, H.; Nishimura, H.; Crofts, N.; Fujita, N.; Sakamoto, W. Amyloplast Membrane Protein SUBSTANDARD STARCH GRAIN6 Controls Starch Grain Size in Rice Endosperm. Plant Physiol. 2016, 170, 1445–1459. [Google Scholar] [CrossRef]
- Fekih, R.; Tamiru, M.; Kanzaki, H.; Abe, A.; Yoshida, K.; Kanzaki, E.; Saitoh, H.; Takagi, H.; Natsume, S.; Undan, J.R.; et al. The rice (Oryza sativa L.) LESION MIMIC RESEMBLING, which encodes an AAA-type ATPase, is implicated in defense response. Mol. Genet. Genom. 2015, 290, 611–622. [Google Scholar] [CrossRef]
- Ouyang, S.Q.; Liu, Y.F.; Liu, P.; Lei, G.; He, S.J.; Ma, B.; Zhang, W.K.; Zhang, J.S.; Chen, S.Y. Receptor-like kinase OsSIK1 improves drought and salt stress tolerance in rice (Oryza sativa) plants. Plant J. 2010, 62, 316–329. [Google Scholar] [CrossRef]
- Dalrymple, D.G. Development of High-Yielding Rice Varieties. In Development and Spread of High-Yielding Rice Varieties in Developing Countries, 2nd ed.; Bureau for Science and Technology Agency for International Development: Washington, DC, USA, 1986; pp. 15–35. [Google Scholar]
- Zeng, D.; Tian, Z.; Rao, Y.; Dong, G.; Yang, Y.; Huang, L.; Leng, Y.; Xu, J.; Sun, C.; Zhang, G.; et al. Rational design of high-yield and superior-quality rice. Nat. Plants 2017, 3, 17031. [Google Scholar] [CrossRef]
- Li, L.; Mao, D.; Prasad, M. Deployment of cold tolerance loci from Oryza sativa ssp. Japonica cv. ‘Nipponbare’ in a high-yielding Indica rice Cultivar ‘93-11’. Plant Breed. 2018, 137, 553–560. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).