The Genome Assembly and Annotation of the Southern Elephant Seal Mirounga leonina


 , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
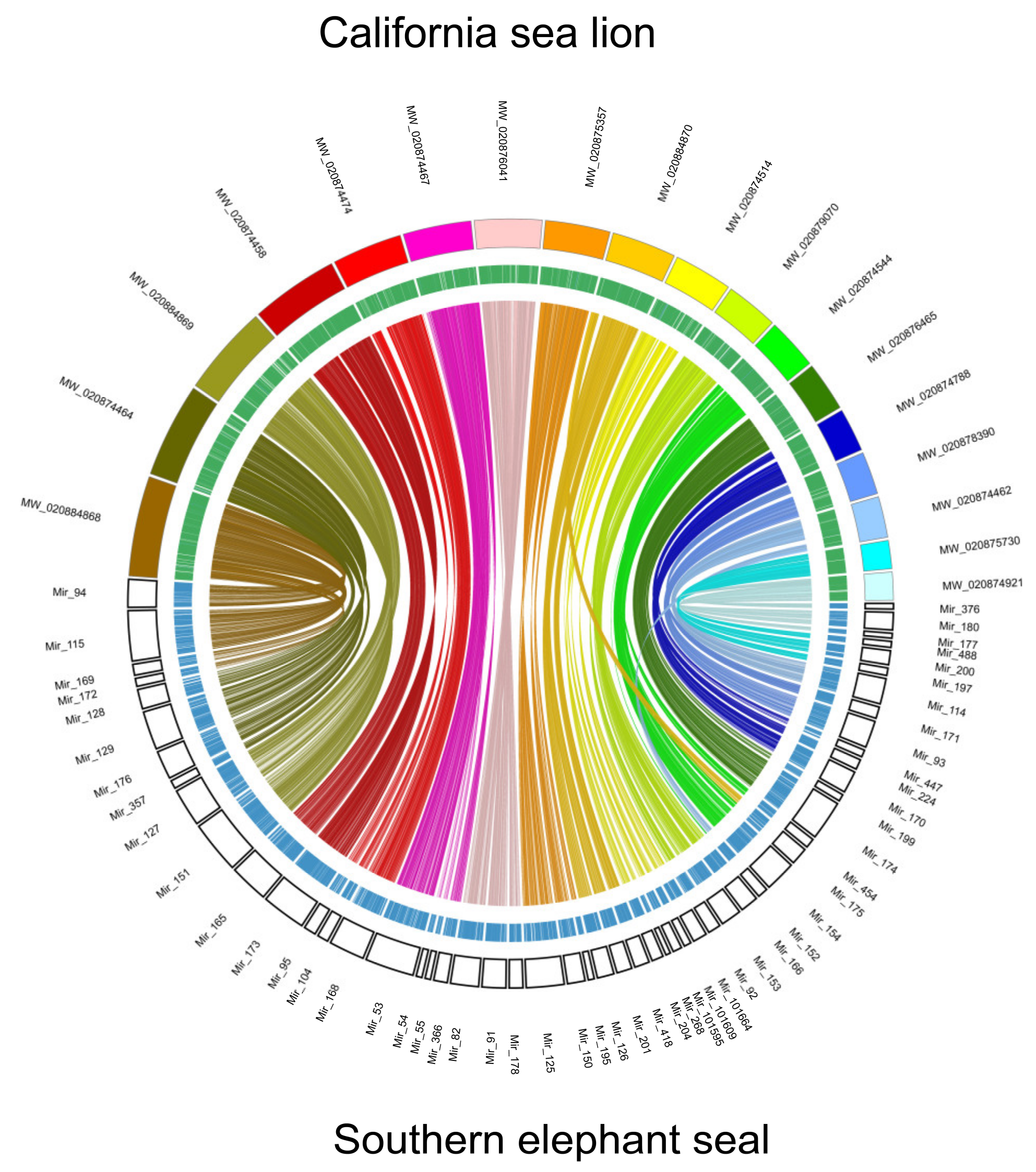
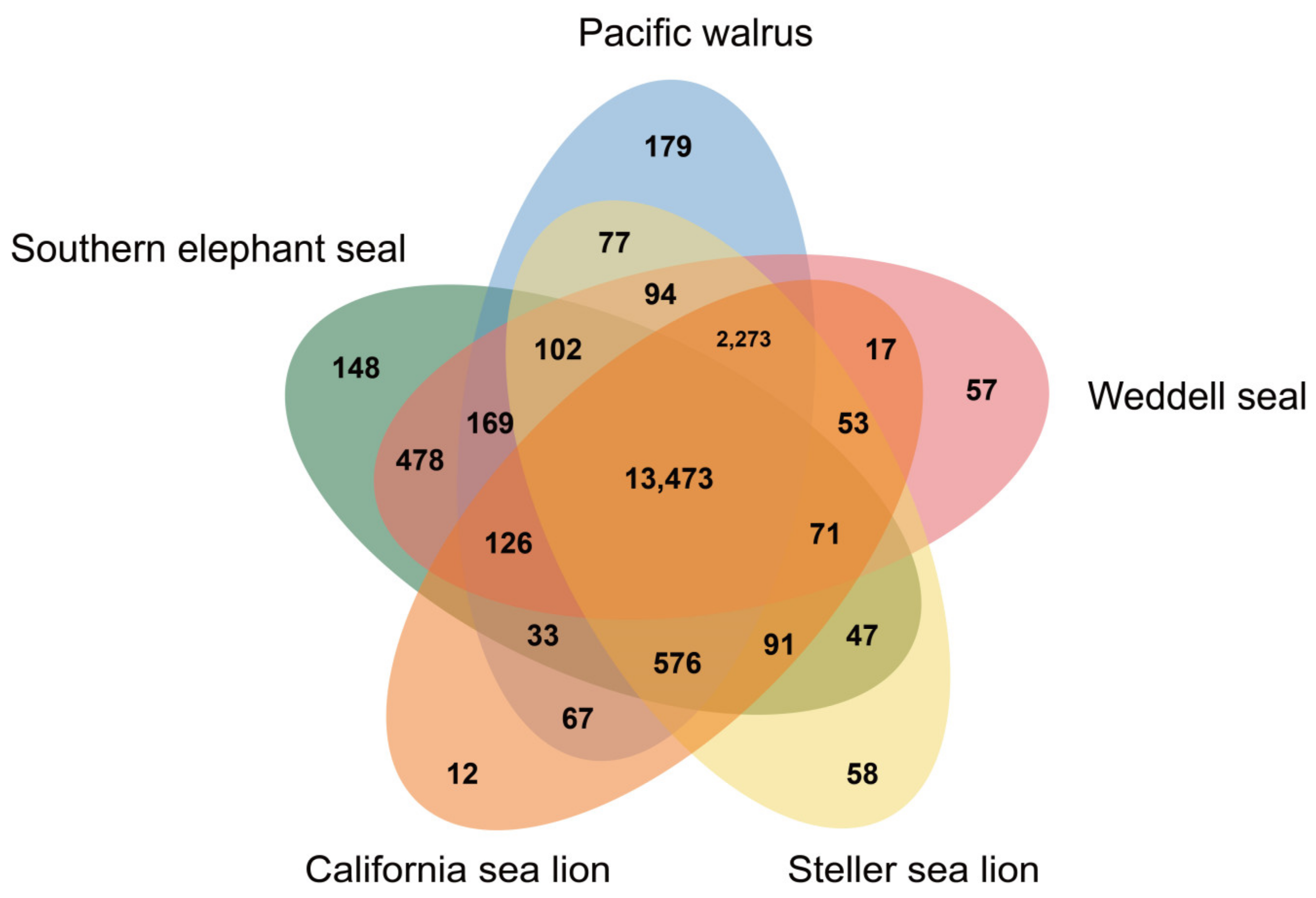
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Le Boeuf, B.J.; Laws, R.M. Elephant Seals: Population Ecology, Behavior, and Physiology; University of California Press: Berkeley, CA, USA, 1994. [Google Scholar]
- Crocker, D.E.; Champagne, C.D.; Fowler, M.A.; Houser, D.S. Adiposity and fat metabolism in lactating and fasting northern elephant seals. Adv. Nutr. 2014, 5, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Rossier, B.C. Osmoregulation during long-term fasting in lungfish and elephant seal: Old and new lessons for the nephrologist. Nephron 2016, 134, 5–9. [Google Scholar] [CrossRef]
- Laws, R.M. Antarctic Seals: Research Methods and Techniques; Cambridge University Press: New York, NY, USA, 1993. [Google Scholar]
- Hofmeyr, G.J.G. Mirounga leonina. In The IUCN Red List of Threatened Species; Species Survival Commission: Gland, Switzerland, 2015. [Google Scholar]
- Biuw, M.; Boehme, L.; Guinet, C.; Hindell, M.; Costa, D.; Charrassin, J.B.; Roquet, F.; Bailleul, F.; Meredith, M.; Thorpe, S.; et al. Variations in behavior and condition of a Southern Ocean top predator in relation to in situ oceanographic conditions. Proc. Natl. Acad. Sci. USA 2007, 104, 13705. [Google Scholar] [CrossRef]
- Kovacs, K.M.; Aguilar, A.; Aurioles, D.; Burkanov, V.; Campagna, C.; Gales, N.; Gelatt, T.; Goldsworthy, S.D.; Goodman, S.J.; Hofmeyr, G.J.; et al. Global threats to pinnipeds. Mar. Mammal Sci. 2012, 28, 414–436. [Google Scholar] [CrossRef]
- Kim, B.-M.; Ahn, D.-H.; Kang, S.; Jeong, J.; Jo, E.; Kim, J.-H.; Rhee, J.-S.; Park, H. De novo Assembly and Annotation of the Blood Transcriptome of the Southern Elephant Seal Mirounga leonina from the South Shetland Islands, Antarctica. Ocean Sci. J. 2019, 54, 307–315. [Google Scholar] [CrossRef]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef]
- Bao, Z.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21, i351–i358. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [PubMed]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.-C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A. The Pfam protein families database in 2019. Nucleic Acids Res. 2018, 47, D427–D432. [Google Scholar] [CrossRef]
- Eddy, S.R.; Mitchison, G.; Durbin, R. Maximum discrimination hidden Markov models of sequence consensus. J. Comput. Biol. 1995, 2, 9–23. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Chalopin, D.; Naville, M.; Plard, F.; Galiana, D.; Volff, J.-N. Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol. Evol. 2015, 7, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Natale, D.A.; Garkavtsev, I.V.; Tatusova, T.A.; Shankavaram, U.T.; Rao, B.S.; Kiryutin, B.; Galperin, M.Y.; Fedorova, N.D.; Koonin, E.V. The COG database: New developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001, 29, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Soderlund, C.; Bomhoff, M.; Nelson, W.M. SyMAP v3.4: A turnkey synteny system with application to plant genomes. Nucleic Acids Res. 2011, 39, e68. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Reads | Total Read Bases | |
|---|---|---|
| Genome | 1,606,582,076 | 242,593,893,476 |
| Transcriptome | 80,958,400 | 19,430,016,000 |
| Assembly | Supernova 2.0 |
|---|---|
| Number of scaffolds | 1115 |
| Total size of scaffolds | 2,417,339,903 |
| Longest scaffolds | 111,625,095 |
| N50 scaffolds length | 54,232,831 |
| Number of scaffolds >10M | 54 |
| Gap (%) | 0.65 |
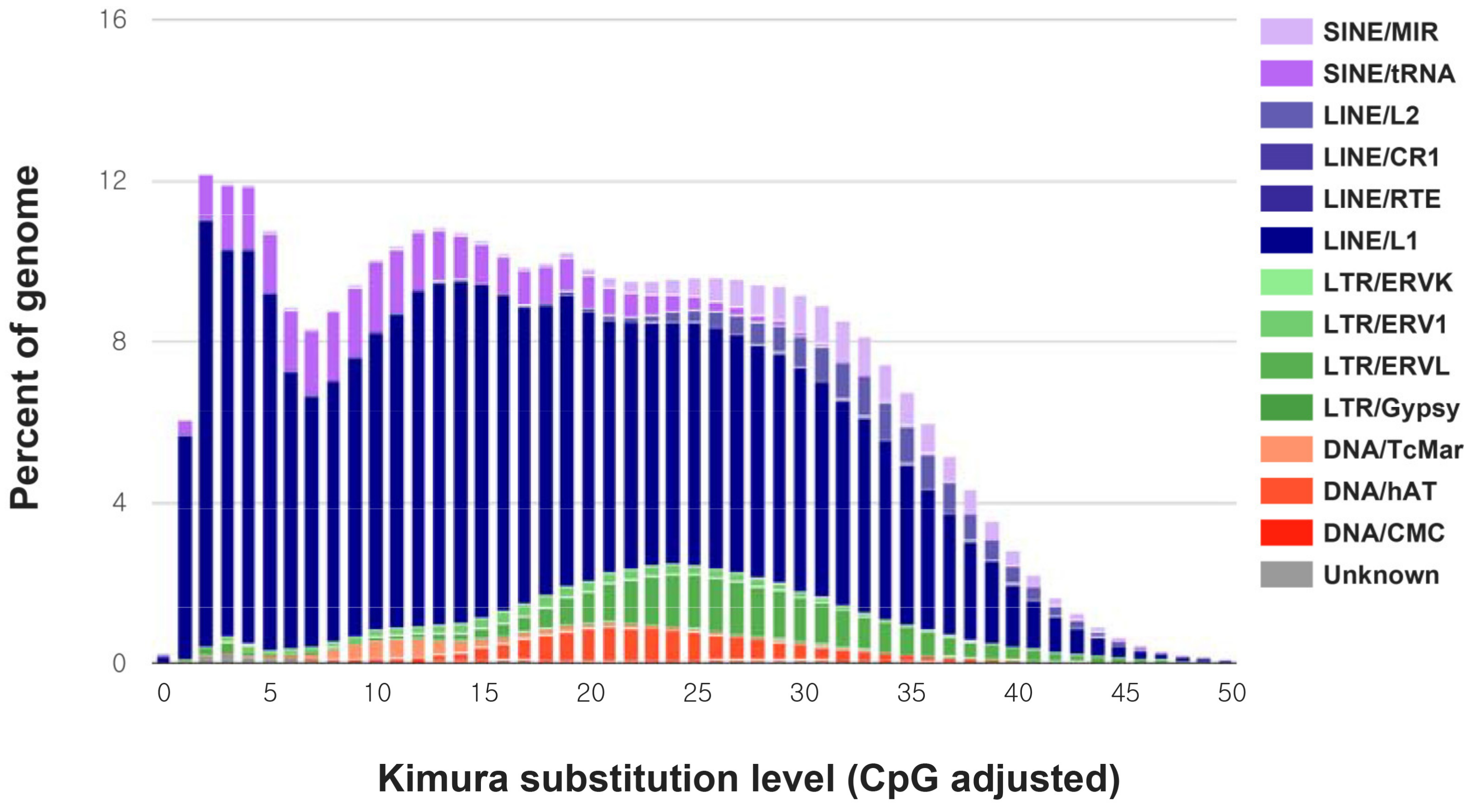
| Number of Elements | Length (bp) | Percentage of Sequence (%) | |
|---|---|---|---|
| SINEs: | 888,301 | 148,028,385 | 6.06 |
| MIRs | 254,250 | 35,222,085 | 1.44 |
| LINEs: | 1,624,363 | 540,626,278 | 22.12 |
| LINE1 | 1,473,069 | 508,962,965 | 20.83 |
| LINE2 | 150,067 | 31,135,376 | 1.27 |
| L3/CR1 | 634 | 81,165 | 0.00 |
| LTR elements: | 268,713 | 74,825,384 | 3.06 |
| ERVL | 81,307 | 29,432,080 | 1.20 |
| ERVL-MaLRs | 134,246 | 29,421,479 | 1.20 |
| ERV_classI | 52,451 | 15,768,764 | 0.65 |
| ERV_classII | 370 | 158,839 | 0.01 |
| DNA elements: | 261,769 | 44,140,597 | 1.81 |
| hAT-Charlie | 146,299 | 24,083,632 | 0.99 |
| TcMar-Tigger | 43,576 | 10,220,361 | 0.42 |
| Unclassified: | 20,630 | 5,747,808 | 0.24 |
| Total interspersed repeats: | 813,368,452 | 33.28 | |
| Small RNA: | 637,161 | 113,111,897 | 4.63 |
| Satellites: | 1867 | 85,913 | 0.00 |
| Simple repeats: | 618,437 | 26,612,557 | 1.09 |
| Low complexity: | 99,311 | 5,218,008 | 0.21 |
| Annotation Database | Annotated Number | Percentage (%) |
|---|---|---|
| No. of genes | 20,457 | |
| nr annotation | 19,901 | 97.3 |
| GO annotation | 3109 | 15.2 |
| KEGG annotation | 11,501 | 56.2 |
| KOG annotation | 15,498 | 75.8 |
| Pfam annotation | 16,733 | 81.8 |
| Swissprot annotation | 19,653 | 96.1 |
| TrEMBL annotation | 17,600 | 86.0 |
| Count | Length Sum (bp) | |
| Exon | 168,375 | 30,497,777 |
| CDS | 167,919 | 28,718,340 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.-M.; Lee, Y.J.; Kim, J.-H.; Kim, J.-H.; Kang, S.; Jo, E.; Lee, S.J.; Lee, J.H.; Chi, Y.M.; Park, H. The Genome Assembly and Annotation of the Southern Elephant Seal Mirounga leonina. Genes 2020, 11, 160. https://doi.org/10.3390/genes11020160
Kim B-M, Lee YJ, Kim J-H, Kim J-H, Kang S, Jo E, Lee SJ, Lee JH, Chi YM, Park H. The Genome Assembly and Annotation of the Southern Elephant Seal Mirounga leonina. Genes. 2020; 11(2):160. https://doi.org/10.3390/genes11020160
Chicago/Turabian StyleKim, Bo-Mi, Yoon Jin Lee, Jeong-Hoon Kim, Jin-Hyoung Kim, Seunghyun Kang, Euna Jo, Seung Jae Lee, Jun Hyuck Lee, Young Min Chi, and Hyun Park. 2020. "The Genome Assembly and Annotation of the Southern Elephant Seal Mirounga leonina" Genes 11, no. 2: 160. https://doi.org/10.3390/genes11020160
APA StyleKim, B.-M., Lee, Y. J., Kim, J.-H., Kim, J.-H., Kang, S., Jo, E., Lee, S. J., Lee, J. H., Chi, Y. M., & Park, H. (2020). The Genome Assembly and Annotation of the Southern Elephant Seal Mirounga leonina. Genes, 11(2), 160. https://doi.org/10.3390/genes11020160