Network Embedding the Protein–Protein Interaction Network for Human Essential Genes Identification


Abstract
1. Introduction
2. Methods
2.1. Bias Random Walk
| Algorithm 1. Bias random walk algorithm. |
| Input G = (V, E, W), Len_walkLists, parameters w, p and q; Output vertex sequence lists: walkLists T = computing transition probabilities (G, p, q, w)//computing transition probabilities for every edge in the network Tnorm = normalizing T by Equation (2) G’ = (V, E, Tnorm) walkLists = {} for iter = 1 to Len_walkLists do for every node u ∈ V do Append u to seq while len(seq) < w: t = seq [-1] // getting the last node of the set seq N(t) = sort (GetNeighbors(t, G’)) // sorting neighbor list of current vertex in alphabetic order n = AliasSampling(N(t), Tnorm) //applying alias sampling with respect to the normalized transition probabilities to select a next visiting neighbor node Append n to seq Append seq to walkLists return walkLists |
2.2. Feature Learning
2.3. Classification
3. Results
3.1. Datasets
3.2. Evaluation Metrics
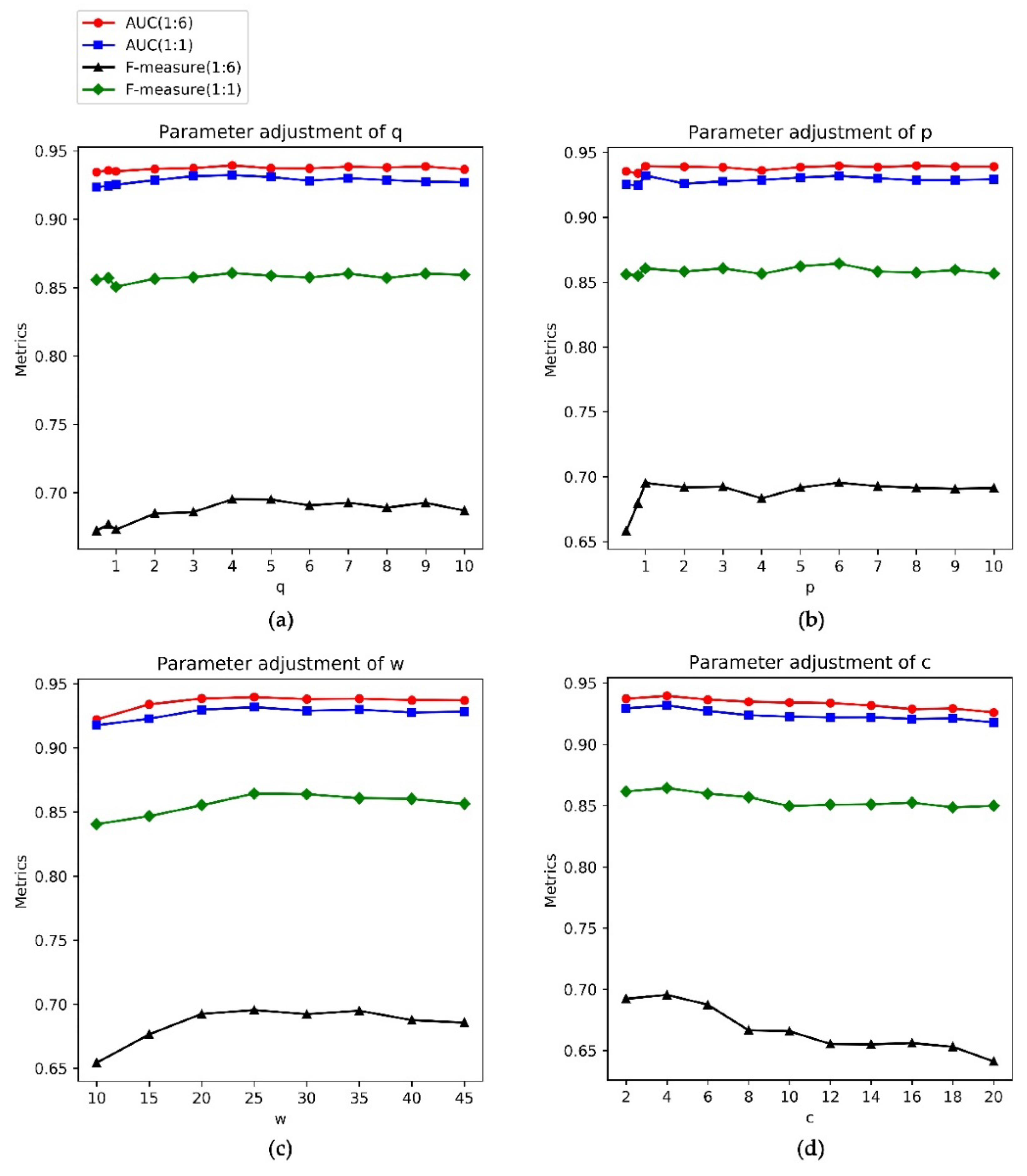
3.3. Parameter Selection
3.4. Comparison with Existing Methods
3.5. Comparison of Different Classifiers
3.6. Feature Representation of Human Essential Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, R.; Lin, Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res. 2009, 37, D455–D458. [Google Scholar] [CrossRef] [PubMed]
- E Clatworthy, A.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Methods 2007, 3, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Giaever, G.; Chu, A.M.; Ni, L.; Connelly, C.; Riles, L.; Véronneau, S.; Dow, S.; Lucau-Danila, A.; Anderson, K.; André, B.; et al. Functional profiling of the Saccharomyces cerevisiae genome. Nat. 2002, 418, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Cullen, L.M.; Arndt, G.M. Genome-wide screening for gene function using RNAi in mammalian cells. Immunol. Cell Boil. 2005, 83, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Roemer, T.; Jiang, B.; Davison, J.; Ketela, T.; Veillette, K.; Breton, A.; Tandia, F.; Linteau, A.; Sillaots, S.; Marta, C.; et al. Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol. Microbiol. 2010, 50, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, D. Understanding protein dispensability through machine-learning analysis of high-throughput data. Bioinform 2005, 21, 575–581. [Google Scholar] [CrossRef]
- Yuan, Y.; Xu, Y.; Xu, J.; Ball, R.L.; Liang, H. Predicting the lethal phenotype of the knockout mouse by integrating comprehensive genomic data. Bioinform. 2012, 28, 1246–1252. [Google Scholar] [CrossRef][Green Version]
- Lloyd, J.P.; Seddon, A.E.; Moghe, G.D.; Simenc, M.C.; Shiu, S.-H. Characteristics of plant essential genes allow for within- and between-species prediction of lethal mutant phenotypes. Plant Cell 2015, 27, 2133–2147. [Google Scholar] [CrossRef]
- Wang, J.; Peng, W.; Wu, F.-X. Computational approaches to predicting essential proteins: A survey. Proteom. Clin. Appl. 2013, 7, 181–192. [Google Scholar] [CrossRef]
- Furney, S.J.; Albà, M.M.; López-Bigas, N. Differences in the evolutionary history of disease genes affected by dominant or recessive mutations. BMC Genom. 2006, 7, 165. [Google Scholar]
- Song, J.; Peng, W.; Wang, F.; Zhang, X.; Tao, L.; Yan, F.; Sung, D.K. An entropy-based method for identifying mutual exclusive driver genes in cancer. IEEE/ACM Trans. Comput. Boil. Bioinform. 2019, 1. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Peng, W.; Wang, F. A random walk-based method to identify driver genes by integrating the subcellular localization and variation frequency into bipartite graph. BMC Bioinform. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Fraser, A. Essential human genes. Cell Syst. 2015, 1, 381–382. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Sci. 2015, 350, 1096–1101. [Google Scholar] [CrossRef]
- Guo, F.-B.; Dong, C.; Hua, H.-L.; Liu, S.; Luo, H.; Zhang, H.-W.; Jin, Y.-T.; Zhang, K.-Y. Accurate prediction of human essential genes using only nucleotide composition and association information. Bioinform. 2017, 33, 1758–1764. [Google Scholar] [CrossRef]
- Jeong, H.; Mason, S.P.; Barabási, A.-L.; Oltvai, Z.N. Lethality and centrality in protein networks. Nat. 2001, 411, 41–42. [Google Scholar] [CrossRef]
- Vallabhajosyula, R.R.; Chakravarti, D.; Lutfeali, S.; Ray, A.; Raval, A. Identifying hubs in protein interaction networks. PloS One 2009, 4, 5344. [Google Scholar] [CrossRef]
- Wuchty, S.; Stadler, P.F. Centers of complex networks. J. Theor. Boil. 2003, 223, 45–53. [Google Scholar] [CrossRef]
- Joy, M.P.; Brock, A.; Ingber, N.E.; Huang, S. High-betweenness proteins in the yeast protein interaction network. J. Biomed. Biotechnol. 2005, 2005, 96–103. [Google Scholar] [CrossRef]
- Stephenson, K.; Zelen, M. Rethinking centrality: Methods and examples. Soc. Networks 1989, 11, 1–37. [Google Scholar] [CrossRef]
- Bonacich, P. Power and centrality: A family of measures. Am. J. Sociol. 1987, 92, 1170–1182. [Google Scholar] [CrossRef]
- Estrada, E.; Rodríguez-Velázquez, J.A. Subgraph centrality in complex networks. Phys. Rev. E 2005, 71, 056103. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, M.; Wang, H.; Pan, Y. Identification of essential proteins based on edge clustering coefficient. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1070–1080. [Google Scholar] [CrossRef]
- Li, M.; Wang, J.; Wang, H.; Pan, Y. Essential proteins discovery from weighted protein interaction networks. Lect. Notes Comput Sc. 2010, 6053, 89–100. [Google Scholar]
- Li, M.; Zhang, H.; Wang, J.-X.; Pan, Y. A new essential protein discovery method based on the integration of protein-protein interaction and gene expression data. BMC Syst. Boil. 2012, 6, 15. [Google Scholar] [CrossRef]
- Tang, X.; Wang, J.; Zhong, J.; Pan, Y. Predicting essential proteins based on weighted degree centrality. IEEE/ACM Trans. Comput. Biol. Bioinf. 2014, 11, 407–418. [Google Scholar] [CrossRef]
- Zhang, F.; Peng, W.; Yang, Y.; Dai, W.; Song, J. A novel method for identifying essential genes by fusing dynamic protein–protein interactive networks. Genes 2019, 10, 31. [Google Scholar] [CrossRef]
- Peng, W.; Wang, J.; Cheng, Y.; Lu, Y.; Wu, F.; Pan, Y. UDoNC: An algorithm for identifying essential proteins based on protein domains and protein-protein interaction networks. IEEE/ACM Trans. Comput. Biol. Bioinf. 2015, 12, 276–288. [Google Scholar] [CrossRef]
- Peng, W.; Wang, J.; Wang, W.; Liu, Q.; Wu, F.-X.; Pan, Y. Iteration method for predicting essential proteins based on orthology and protein-protein interaction networks. BMC Syst. Boil. 2012, 6, 87. [Google Scholar] [CrossRef]
- Zhong, J.; Sun, Y.; Peng, W.; Xie, M.; Yang, J.; Tang, X. XGBFEMF: An XGBoost-based framework for essential protein prediction. IEEE Trans. NanoBioscience 2018, 17, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Li, M.; Chen, L.; Wang, L. Predicting protein functions by using unbalanced random walk algorithm on three biological networks. IEEE/ACM Trans. Comput. Biol. Bioinf. 2017, 14, 360. [Google Scholar] [CrossRef] [PubMed]
- Mikolov, T.; Sutskever, I.; Chen, K.; Corrado, G.; Dean, J. Distributed Representations of Words and Phrases and their Compositionality. In Proceedings of the International Conference on Neural Information Processing Systems, Lake Tahoe, NV, USA, 5–8 December 2013. [Google Scholar]
- Perozzi, B.; Al-Rfou, R.; Skiena, S. DeepWalk: Online Learning of Social Representations. In Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, New York, NY, USA, 24–27 August 2014; ACM Digital Library: New York, NY, USA, 2014; pp. 701–710. [Google Scholar]
- Tang, J.; Qu, M.; Wang, M.; Zhang, M.; Yan, J.; Mei, Q. LINE: Large-scale information network embedding. In Proceedings of the 24th International Conference on World Wide Web, WWW 2015, Florence, Italy, 18–22 May 2015. [Google Scholar]
- Wang, Y.; Yao, Y.; Tong, H.; Xu, F.; Lu, J. A brief review of network embedding. Big Data Min. Anal. 2019, 2, 35–47. [Google Scholar] [CrossRef]
- Ye, Z.; Zhao, H.; Zhang, K.; Wang, Z.; Zhu, Y. Network representation based on the joint learning of three feature views. Big Data Min. Anal. 2019, 2, 248–260. [Google Scholar] [CrossRef]
- Grover, A.; Leskovec, J. node2vec: Scalable feature learning for networks. KDD 2016, 855–864. [Google Scholar]
- Dai, W.; Chang, Q.; Peng, W.; Zhong, J.; Li, Y. Identifying human essential genes by network embedding protein-protein interaction network. Lect. Notes Comput Sc. 2019, 11490, 127–137. [Google Scholar]
- Wu, J.; Zhang, Q.; Wu, W.; Pang, T.; Hu, H.; Chan, W.K.B.; Ke, X.; Zhang, Y. WDL-RF: Predicting bioactivities of ligand molecules acting with G protein-coupled receptors by combining weighted deep learning and random forest. Bioinform. 2018, 34, 2271–2282. [Google Scholar] [CrossRef]
- Acencio, M.L.; Lemke, N. Towards the prediction of essential genes by integration of network topology, cellular localization and biological process information. BMC Bioinform. 2009, 10, 290. [Google Scholar] [CrossRef]
- Liao, J.; Chin, K.-V. Logistic regression for disease classification using microarray data: Model selection in a large p and small n case. Bioinform. 2007, 23, 1945–1951. [Google Scholar] [CrossRef]
- Chou, K.C.; Shen, H.B. Predicting eukaryotic protein subcellular location by fusing optimized evidence-theoretic K-Nearest Neighbor classifiers. J. Proteome. Res. 2006, 5, 1888–1897. [Google Scholar] [CrossRef]
- Cheng, J.; Xu, Z.; Wu, W.; Zhao, L.; Li, X.; Liu, Y.; Tao, S. Training set selection for the prediction of essential genes. PloS One 2014, 9, e86805. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Feng, X.; Stein, L. A human functional protein interaction network and its application to cancer data analysis. Genome Boil. 2010, 11, R53. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wernersson, R.; Hansen, R.B.; Horn, H.; Mercer, J.; Slodkowicz, G.; Workman, C.T.; Rigina, O.; Rapacki, K.; Stærfeldt, H.H.; et al. A scored human protein–protein interaction network to catalyze genomic interpretation. Nat. Methods 2016, 14, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, M.; Wang, J.; Pan, Y.; Wu, F.-X. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosyst. 2015, 127, 67–72. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data set | Genes | Interactions | Essential genes | Training and testing genes |
|---|---|---|---|---|
| FIs | 12277 | 230243 | 1359 | 6747 |
| InWeb_IM | 17428 | 625641 | 1512 | 10548 |
| Methods | Precision | Recall | SP | NPV | F-measure | MCC | ACC | AUC | AP |
|---|---|---|---|---|---|---|---|---|---|
| DeepWalk (1:4) | 0.771 | 0.621 | 0.954 | 0.909 | 0.688 | 0.625 | 0.887 | 0.905 | 0.734 |
| LINE (1:4) | 0.837 | 0.394 | 0.981 | 0.866 | 0.539 | 0.513 | 0.863 | 0.856 | 0.693 |
| Centrality (1:4) | 0.608 | 0.523 | 0.917 | 0.879 | 0.562 | 0.464 | 0.836 | 0.753 | 0.550 |
| Z-curve (1:4) | 0.526 | 0.530 | 0.880 | 0.882 | 0.528 | 0.409 | 0.810 | 0.834 | 0.522 |
| Our method (1:4) | 0.822 | 0.597 | 0.968 | 0.905 | 0.692 | 0.641 | 0.893 | 0.913 | 0.769 |
| DeepWalk (1:1) | 0.833 | 0.846 | 0.830 | 0.843 | 0.839 | 0.676 | 0.838 | 0.907 | 0.896 |
| LINE (1:1) | 0.705 | 0.847 | 0.647 | 0.808 | 0.770 | 0.503 | 0.747 | 0.839 | 0.848 |
| Centrality (1:1) | 0.852 | 0.553 | 0.904 | 0.669 | 0.671 | 0.488 | 0.728 | 0.760 | 0.797 |
| Z-curve (1:1) | 0.733 | 0.800 | 0.708 | 0.780 | 0.765 | 0.511 | 0.754 | 0.824 | 0.783 |
| Our method (1:1) | 0.859 | 0.836 | 0.863 | 0.840 | 0.847 | 0.699 | 0.849 | 0.914 | 0.902 |
| Methods | Precision | Recall | SP | NPV | F-measure | MCC | ACC | AUC | AP |
|---|---|---|---|---|---|---|---|---|---|
| DeepWalk (1:6) | 0.749 | 0.515 | 0.971 | 0.923 | 0.610 | 0.571 | 0.906 | 0.904 | 0.667 |
| LINE (1:6) | 0.819 | 0.165 | 0.994 | 0.877 | 0.275 | 0.332 | 0.875 | 0.832 | 0.547 |
| Centrality (1:6) | 0.527 | 0.562 | 0.916 | 0.926 | 0.544 | 0.465 | 0.865 | 0.828 | 0.511 |
| Z-curve (1:6) | 0.485 | 0.462 | 0.918 | 0.911 | 0.473 | 0.388 | 0.853 | 0.840 | 0.456 |
| Our method (1:6) | 0.841 | 0.550 | 0.983 | 0.929 | 0.665 | 0.641 | 0.921 | 0.915 | 0.762 |
| DeepWalk (1:1) | 0.817 | 0.848 | 0.811 | 0.842 | 0.832 | 0.659 | 0.829 | 0.908 | 0.894 |
| LINE (1:1) | 0.641 | 0.905 | 0.492 | 0.839 | 0.750 | 0.436 | 0.699 | 0.804 | 0.792 |
| Centrality (1:1) | 0.816 | 0.713 | 0.839 | 0.745 | 0.761 | 0.557 | 0.776 | 0.851 | 0.841 |
| Z-curve (1:1) | 0.730 | 0.777 | 0.713 | 0.762 | 0.753 | 0.491 | 0.745 | 0.827 | 0.801 |
| Our method (1:1) | 0.855 | 0.858 | 0.854 | 0.858 | 0.857 | 0.713 | 0.856 | 0.928 | 0.921 |
| Methods | Precision | Recall | SP | NPV | F-measure | MCC | Accuracy | AUC | AP |
|---|---|---|---|---|---|---|---|---|---|
| DNN (1 layer, 1:4) | 0.258 | 0.770 | 0.220 | 0.805 | 0.234 | 0.026 | 0.667 | 0.460 | 0.242 |
| DNN (3 layers, 1:4) | 0.248 | 0.407 | 0.690 | 0.822 | 0.308 | 0.083 | 0.633 | 0.534 | 0.245 |
| DT (1:4) | 0.623 | 0.619 | 0.906 | 0.904 | 0.621 | 0.526 | 0.848 | 0.763 | 0.660 |
| NB (1:4) | 0.553 | 0.737 | 0.850 | 0.928 | 0.632 | 0.531 | 0.827 | 0.880 | 0.683 |
| KNN (1:4) | 0.795 | 0.628 | 0.959 | 0.911 | 0.702 | 0.644 | 0.893 | 0.889 | 0.782 |
| LR (1:4) | 0.787 | 0.585 | 0.960 | 0.902 | 0.671 | 0.613 | 0.885 | 0.914 | 0.755 |
| SVM (1:4) | 0.822 | 0.597 | 0.968 | 0.905 | 0.692 | 0.641 | 0.893 | 0.913 | 0.769 |
| RF (1:4) | 0.826 | 0.646 | 0.966 | 0.916 | 0.726 | 0.674 | 0.902 | 0.927 | 0.799 |
| ET (1:4) | 0.828 | 0.648 | 0.966 | 0.916 | 0.727 | 0.676 | 0.902 | 0.932 | 0.806 |
| DNN (1 layer, 1:1) | 0.554 | 0.452 | 0.503 | 0.504 | 0.527 | 0.007 | 0.503 | 0.500 | 0.519 |
| DNN (3 layers, 1:1) | 0.535 | 0.538 | 0.532 | 0.536 | 0.536 | 0.070 | 0.535 | 0.562 | 0.553 |
| DT (1:1) | 0.768 | 0.788 | 0.762 | 0.783 | 0.778 | 0.551 | 0.775 | 0.775 | 0.831 |
| NB (1:1) | 0.822 | 0.765 | 0.834 | 0.780 | 0.792 | 0.600 | 0.799 | 0.876 | 0.873 |
| KNN (1:1) | 0.837 | 0.805 | 0.844 | 0.812 | 0.821 | 0.649 | 0.824 | 0.895 | 0.906 |
| LR (1:1) | 0.839 | 0.827 | 0.841 | 0.829 | 0.833 | 0.668 | 0.834 | 0.910 | 0.907 |
| SVM (1:1) | 0.859 | 0.836 | 0.863 | 0.840 | 0.847 | 0.699 | 0.849 | 0.914 | 0.902 |
| RF (1:1) | 0.859 | 0.844 | 0.861 | 0.846 | 0.851 | 0.705 | 0.852 | 0.921 | 0.921 |
| ET (1:1) | 0.867 | 0.840 | 0.872 | 0.845 | 0.853 | 0.712 | 0.856 | 0.923 | 0.922 |
| Methods | Precision | Recall | SP | NPV | F-measure | MCC | Accuracy | AUC | AP |
|---|---|---|---|---|---|---|---|---|---|
| DNN (1 layer, 1:6) | 0.355 | 0.772 | 0.206 | 0.877 | 0.261 | 0.103 | 0.712 | 0.499 | 0.206 |
| DNN (3 layers, 1:6) | 0.350 | 0.255 | 0.921 | 0.881 | 0.295 | 0.202 | 0.826 | 0.483 | 0.221 |
| DT (1:6) | 0.584 | 0.588 | 0.930 | 0.931 | 0.586 | 0.517 | 0.881 | 0.759 | 0.616 |
| NB (1:6) | 0.456 | 0.697 | 0.861 | 0.945 | 0.551 | 0.473 | 0.837 | 0.877 | 0.615 |
| KNN (1:6) | 0.778 | 0.564 | 0.973 | 0.930 | 0.654 | 0.617 | 0.914 | 0.888 | 0.740 |
| LR (1:6) | 0.785 | 0.591 | 0.973 | 0.934 | 0.675 | 0.637 | 0.918 | 0.931 | 0.749 |
| SVM (1:6) | 0.841 | 0.550 | 0.983 | 0.929 | 0.665 | 0.641 | 0.921 | 0.915 | 0.762 |
| RF (1:6) | 0.799 | 0.615 | 0.974 | 0.938 | 0.695 | 0.659 | 0.923 | 0.940 | 0.776 |
| ET (1:6) | 0.816 | 0.600 | 0.977 | 0.936 | 0.692 | 0.659 | 0.925 | 0.943 | 0.779 |
| DNN (1 layer, 1:1) | 0.652 | 0.504 | 0.568 | 0.591 | 0.607 | 0.157 | 0.578 | 0.603 | 0.640 |
| DNN (3 layers, 1:1) | 0.737 | 0.497 | 0.823 | 0.620 | 0.593 | 0.338 | 0.659 | 0.637 | 0.692 |
| DT (1:1) | 0.802 | 0.791 | 0.805 | 0.794 | 0.797 | 0.596 | 0.798 | 0.798 | 0.849 |
| NB (1:1) | 0.836 | 0.708 | 0.862 | 0.747 | 0.767 | 0.576 | 0.785 | 0.874 | 0.872 |
| KNN (1:1) | 0.853 | 0.843 | 0.854 | 0.845 | 0.848 | 0.697 | 0.849 | 0.904 | 0.906 |
| LR (1:1) | 0.865 | 0.834 | 0.870 | 0.840 | 0.849 | 0.704 | 0.852 | 0.925 | 0.920 |
| SVM (1:1) | 0.855 | 0.858 | 0.854 | 0.858 | 0.857 | 0.713 | 0.856 | 0.928 | 0.921 |
| RF (1:1) | 0.844 | 0.886 | 0.836 | 0.880 | 0.864 | 0.723 | 0.861 | 0.932 | 0.920 |
| ET (1:1) | 0.853 | 0.879 | 0.849 | 0.876 | 0.866 | 0.729 | 0.864 | 0.934 | 0.928 |
| Data set | DC | BC | CC | NC | IC |
|---|---|---|---|---|---|
| FIs | 0.9262 | 0.8040 | 0.9998 | 0.9911 | 0.9794 |
| InWeb_IM | 0.9617 | 0.8372 | 0.9999 | 0.9938 | 0.9839 |
| Data Set | Features | Max Size | Min Size | Median Size | Silhouette | Dunn | Avg(-log(p-value)) |
|---|---|---|---|---|---|---|---|
| FIs | Feature representation | 289 | 21 | 51.5 | 0.3242 | 0.59 | 63.50 |
| Connection relationship | 902 | 6 | 19.5 | 0.2448 | 0.58 | 45.01 | |
| InWeb_IM | Feature representation | 176 | 20 | 61.5 | 0.2279 | 0.63 | 54.95 |
| Connection relationship | 1022 | 1 | 13 | 0.1619 | 0.25 | 30.11 |
| GO ID | Description | p-value | Genes in Cluster | Gene Ratio |
|---|---|---|---|---|
| GO:0000377 | RNA splicing, via transesterification reactions with bulged adenosine as nucleophile | 6.64·10−191 | PLRG1/PABPN1/SNRNP35/HNRNPM/RBMX/RBM22/DHX9/MAGOH/XAB2/SRRM2/SNRPF/SMNDC1/SRRM1/SF3B5/PPIE/CTNNBL1/PRPF40A/SNRNP70/PRPF4B/EIF4A3/PRPF19/BUD31/HNRNPL/NCBP1/DHX8/SNRPC/CWC22/CPSF1/RNPC3/HNRNPC/TFIP11/SNU13/CLP1/SNRNP200/ISY1/TXNL4A/CPSF2/USP39/SNRNP27/ALYREF/DDX23/CPSF3/PCBP1/DDX39B/PCF11/SYMPK/SF3B1/BCAS2/SRSF1/SF3A2/WDR33/SRSF11/DHX38/SNRNP25/SNRPE/HNRNPH1/ZMAT5/RBM17/SNRNP48/CHERP/PUF60/FIP1L1/HNRNPK/HNRNPA2B1/CSTF3/LSM7/CDC40/SNRPD3/PRPF3/PRPF31/NUDT21/SART3/AQR/CRNKL1/U2AF2/PPWD1/PRPF6/SRSF2/LSM4/SRRT/DHX16/SART1/CSTF1/SF1/SRSF7/SNRPB/DHX15/EFTUD2/NCBP2/PRPF4/SF3B2/SLU7/CDC5L/SF3A3/LSM2/GPKOW/SUGP1/HNRNPU/SF3B4/CPSF4/PRPF8/SRSF3/SNRPD2/HSPA8/SNW1/U2SURP/DDX46/SF3A1/SF3B3/PDCD7 | 110/116 |
| GO:0070125 | mitochondrial translational elongation | 6.97·10−158 | MRPL28/MRPL22/MRPL17/MRPL48/MRPL37/MRPL4/MRPL47/TUFM/MRPL23/MRPL9/MRPL24/MRPS12/MRPL11/MRPL10/MRPL38/ERAL1/MRPL46/MRPS6/MRPS25/MRPL41/MRPS27/MRPL12/MRPS16/MRPS23/MRPL34/MRPS34/MRPL43/MRPL15/MRPS24/MRPL35/MRPL40/MRPL57/MRPL21/GFM1/MRPS7/PTCD3/MRPS22/MRPL13/MRPL51/MRPL53/MRPS2/MRPL14/MRPS5/MRPL45/MRPL18/DAP3/AURKAIP1/MRPL19/MRPS15/MRPL20/MRPL39/MRPL44/GADD45GIP1/MRPS18A/MRPS14/MRPS11/MRPS31/MRPS18C/MRPS18B/MRPS30/MRPL16/MRPS35/MRPS10/MRPL33 | 64/67 |
| GO:0000184 | nuclear-transcribed mRNA catabolic process, nonsense-mediated decay | 7.98·10−107 | UPF1/RPS16/RPSA/SMG5/RPS14/RPL10A/RPL8/RPS11/SMG7/RPL13A/RPLP1/ETF1/RPL27A/EIF4G1/GSPT1/RPLP2/RPL4/RPS13/RPL18/RPS4X/RPL36/RPS10/RPL23A/RPS12/RPS5/RPL9/SMG6/RPS18/RPS21/RPS7/RPL29/RPL31/RPL12/RPL10/RPL7/PABPC1/RPL3/RPS15A/RPL37A/RPL18A/RPL19/RPS25/SMG1/RPL11/RPL7A/RPS15/RPS9/RPS2/UPF2/RPL27/RPL13/RPL14/RPL15/RPS29/RPL38/RPLP0/RPS8/RPS6 | 58/96 |
| GO:0000819 | sister chromatid segregation | 1.97·10−94 | PMF1/NSL1/BUB3/ESPL1/NUP43/BIRC5/SMC2/XPO1/CENPC/NUF2/KIF18A/SEH1L/SKA1/NUP62/SEC13/NCAPD3/NCAPD2/TTK/MAU2/SMC5/NSMCE2/CENPA/CDCA8/RAD21/AHCTF1/CENPM/RANGAP1/SPC24/PLK1/SMC1A/AURKB/CENPK/INCENP/RANBP2/NUP85/SPC25/CKAP5/NUP107/CENPE/SMC3/NUP98/CENPL/PAFAH1B1/MAD2L1/CENPN/SPDL1/NDC80/CDC20/DSN1/BUB1B/NUP133/TPR/KPNB1/CENPI/SMC4/NUDC/SGO1/CCNB1/RAN/NUP160/CDCA5 | 61/91 |
| GO:0006364 | rRNA processing | 5.08·10−94 | WDR75/BMS1/EXOSC9/UTP6/NOP56/HEATR1/EXOSC1/UTP20/UTP4/UTP14A/EXOSC8/RRP36/NOL6/MPHOSPH10/DDX47/RRP9/TBL3/PWP2/EXOSC7/WDR3/WDR18/IMP4/EXOSC5/EXOSC4/WDR46/UTP18/DIEXF/UTP11/LAS1L/UTP3/UTP15/NOC4L/FBL/IMP3/NOP14/PDCD11/TEX10/NOP58/RCL1/XRN2/KRR1/DDX49/EXOSC3/EXOSC10/WDR43/DDX52/EXOSC6/NOL11/EXOSC2/WDR36 | 50/50 |
| GO:0031145 | anaphase-promoting complex-dependent catabolic process | 5.71·10−89 | ANAPC11/PSMD7/PSMD14/PSMA7/PSMB3/PSMB6/PSMA3/PSMD12/PSMA4/PSMB5/PSMA2/ANAPC2/PSMB1/ANAPC4/PSMD6/ANAPC5/PSMA5/PSMD2/PSMD4/PSMC6/PSMC1/CDC16/ANAPC15/PSMD1/PSMD11/PSMB4/PSMD3/PSMD8/CUL3/ANAPC10/PSMC4/PSMA1/PSMC2/PSMC3/PSMB7/AURKA/PSMC5/PSMD13/PSMB2/CDC23 | 40/51 |
| GO:0098781 | ncRNA transcription | 2.19·10−81 | GTF2E1/PHAX/INTS5/TAF13/TAF6/POLR2E/ZC3H8/ELL/POLR2H/TAF8/INTS7/POLR2F/BRF1/GTF2A1/POLR2B/SNAPC2/POLR2I/GTF3C2/INTS9/TAF5/INTS8/GTF2A2/INTS2/POLR2G/POLR2C/GTF3C4/INTS3/SNAPC4/GTF3C3/GTF2B/POLR2D/INTS6/SNAPC3/INTS1/CCNK/CDK9/ICE1/POLR2L/RPAP2/GTF3C5/INTS4/CDK7/GTF2E2/GTF3C1 | 44/80 |
| GO:0006270 | DNA replication initiation | 2.12·10−66 | CDC7/MCM2/POLA1/MCM4/POLE2/ORC5/ORC2/CDC45/MCM3/ORC4/MCM7/ORC3/MCM10/CDC6/MCM6/ORC6/ORC1/MCM5/PRIM1/POLA2/GINS2/CDT1/CDK2/GINS4/POLE | 25/28 |
| GO:0048193 | Golgi vesicle transport | 1.46·10−61 | GBF1/TRAPPC1/VPS52/COG4/KIF11/COPB1/DCTN5/VPS54/YKT6/DCTN6/STX18/COPA/COPB2/NSF/COG8/PREB/TMED2/TMED10/TRAPPC4/DCTN4/KIF23/COPG1/TRAPPC5/ARFRP1/ARCN1/NAPA/COPE/TRAPPC8/RINT1/SCFD1/COPZ1/STX5/TRAPPC11/SYS1/NBAS/COG3/SEC16A/TRAPPC3/RACGAP1/GOSR2 | 40/ 47 |
| GO:0042254 | ribosome biogenesis | 5.70·10−53 | WDR12/FTSJ3/NOP2/DDX56/MAK16/BRIX1/RRP1/NAT10/ABCE1/GTPBP4/PPAN/MDN1/PES1/EBNA1BP2/DIS3/NOP16/POP4/DDX27/EFL1/SDAD1/NSA2/HEATR3/RPF1/RSL1D1/RRS1/TSR2/TRMT112/LSG1/NHP2/NOP10/NOL9/DKC1/NIP7/GNL2/ISG20L2/RPL7L1/SURF6/DDX51/RRP15/PELP1/NGDN/NOC2L/WDR74 | 43/ 81 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, W.; Chang, Q.; Peng, W.; Zhong, J.; Li, Y. Network Embedding the Protein–Protein Interaction Network for Human Essential Genes Identification. Genes 2020, 11, 153. https://doi.org/10.3390/genes11020153
Dai W, Chang Q, Peng W, Zhong J, Li Y. Network Embedding the Protein–Protein Interaction Network for Human Essential Genes Identification. Genes. 2020; 11(2):153. https://doi.org/10.3390/genes11020153
Chicago/Turabian StyleDai, Wei, Qi Chang, Wei Peng, Jiancheng Zhong, and Yongjiang Li. 2020. "Network Embedding the Protein–Protein Interaction Network for Human Essential Genes Identification" Genes 11, no. 2: 153. https://doi.org/10.3390/genes11020153
APA StyleDai, W., Chang, Q., Peng, W., Zhong, J., & Li, Y. (2020). Network Embedding the Protein–Protein Interaction Network for Human Essential Genes Identification. Genes, 11(2), 153. https://doi.org/10.3390/genes11020153