Unraveling the Deep Genetic Architecture for Seedlessness in Grapevine and the Development and Validation of a New Set of Markers for VviAGL11-Based Gene-Assisted Selection

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Seedlessness Phenotypic Evaluation for QTL Mapping
2.3. Seedlessness Phenotypic Evaluation for Validation Purposes
2.4. Genetic Characterization of MA × CS by Illumina Vitis 20k SNP Chip Genotyping
2.5. Fine QTL Analysis of Seedless Subtraits in Muscat of Alexandria × Crimson Seedless F1 Mapping Population
2.6. Identification of Putative Candidate Genes within QTL Confidence Intervals
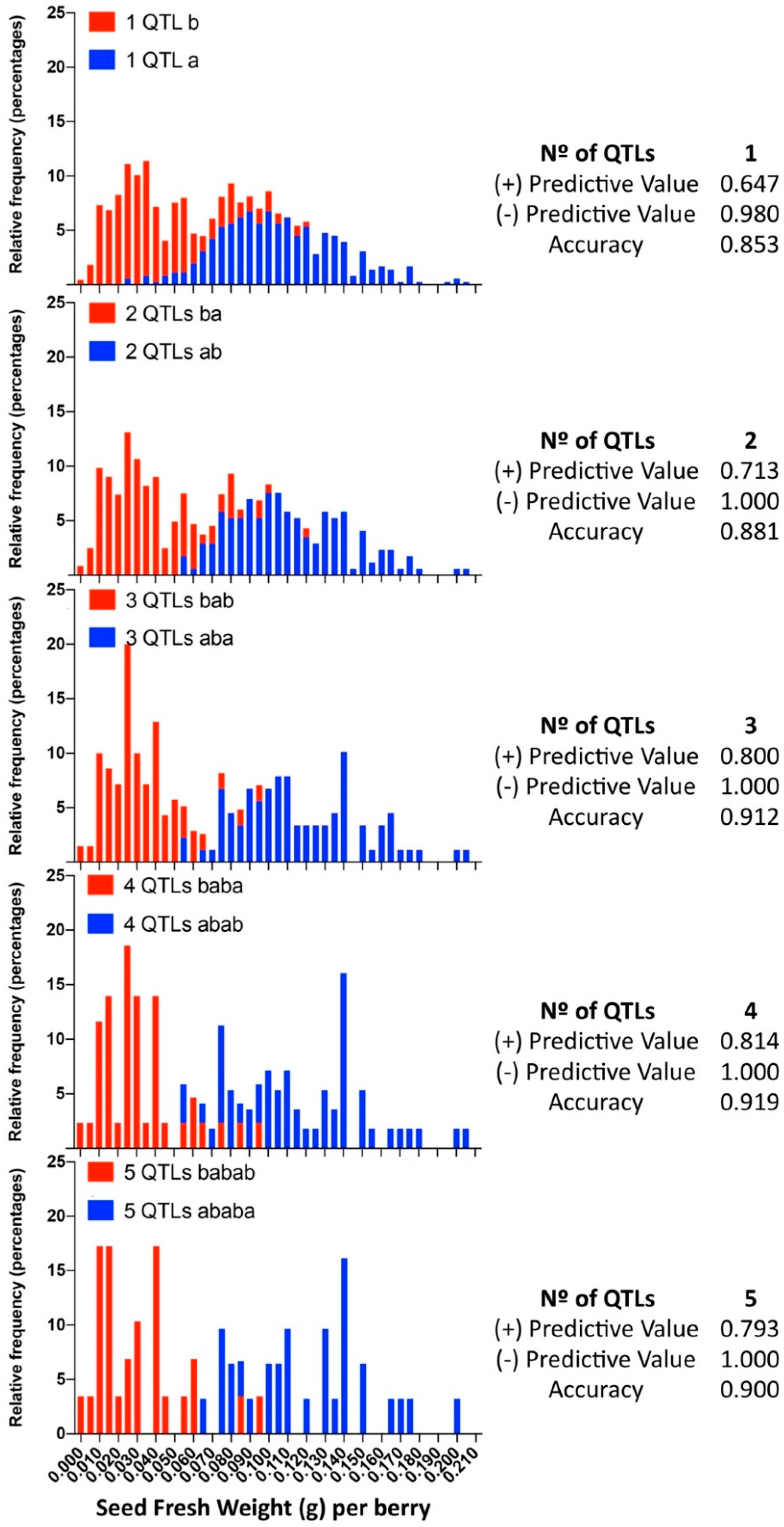
2.7. Evaluation of Multi-QTL Predictive Model to Increase Assisted Selection Efficiency
2.8. Sequencing of VviAGL11 Regulatory and 5′UTR Regions for Variant Discovery and the Design of a New SSR For Assisted Selection
2.9. PCR Validation Assays of SSR 5U_VviAGL11
2.10. Real-Time PCR and High-Resolution Melt Analysis for Rapid Detection of the Substitution Arg-197-Leu in the Seedless Allele of VviAGL11
2.11. Tetra-Primer Amplification Refractory Mutation System PCR for Non-Sophisticated Detection of the Seedless Allele
2.12. Validation Assay: Comparison between SSR p3_VvAGL11, SNP e7_VviAGL11, and SSR 5U_VviAGL11
3. Results
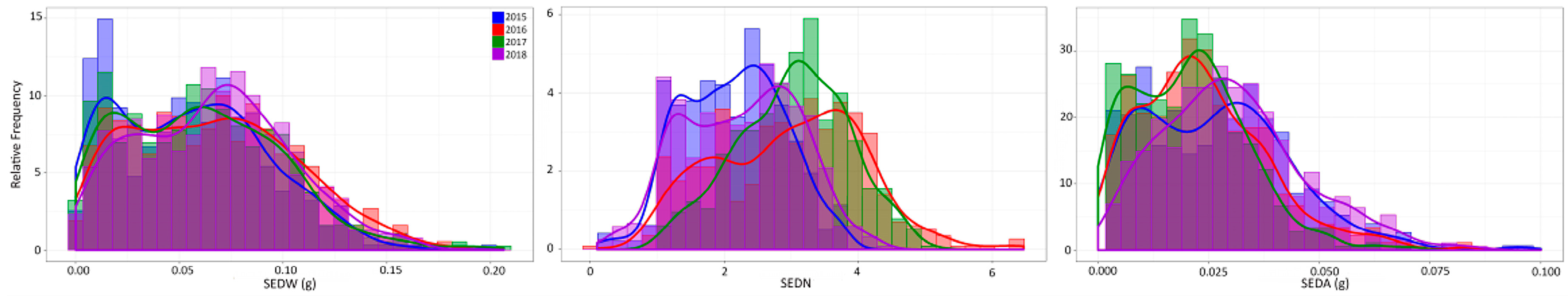
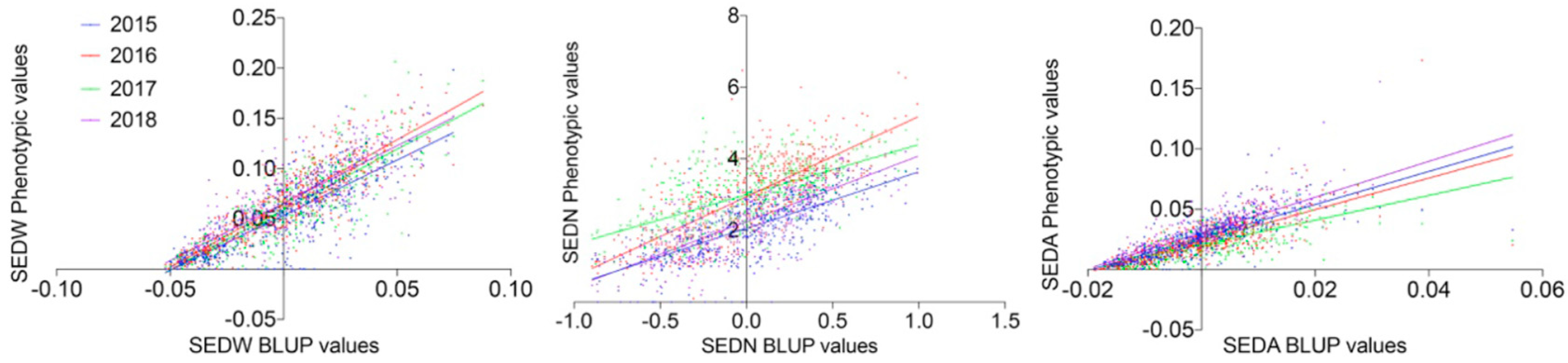
3.1. Phenotypic Evaluations of the MA × CS Progeny Across Four Consecutive Seasons
3.2. Genetic Map Construction for the MA × CS Progeny
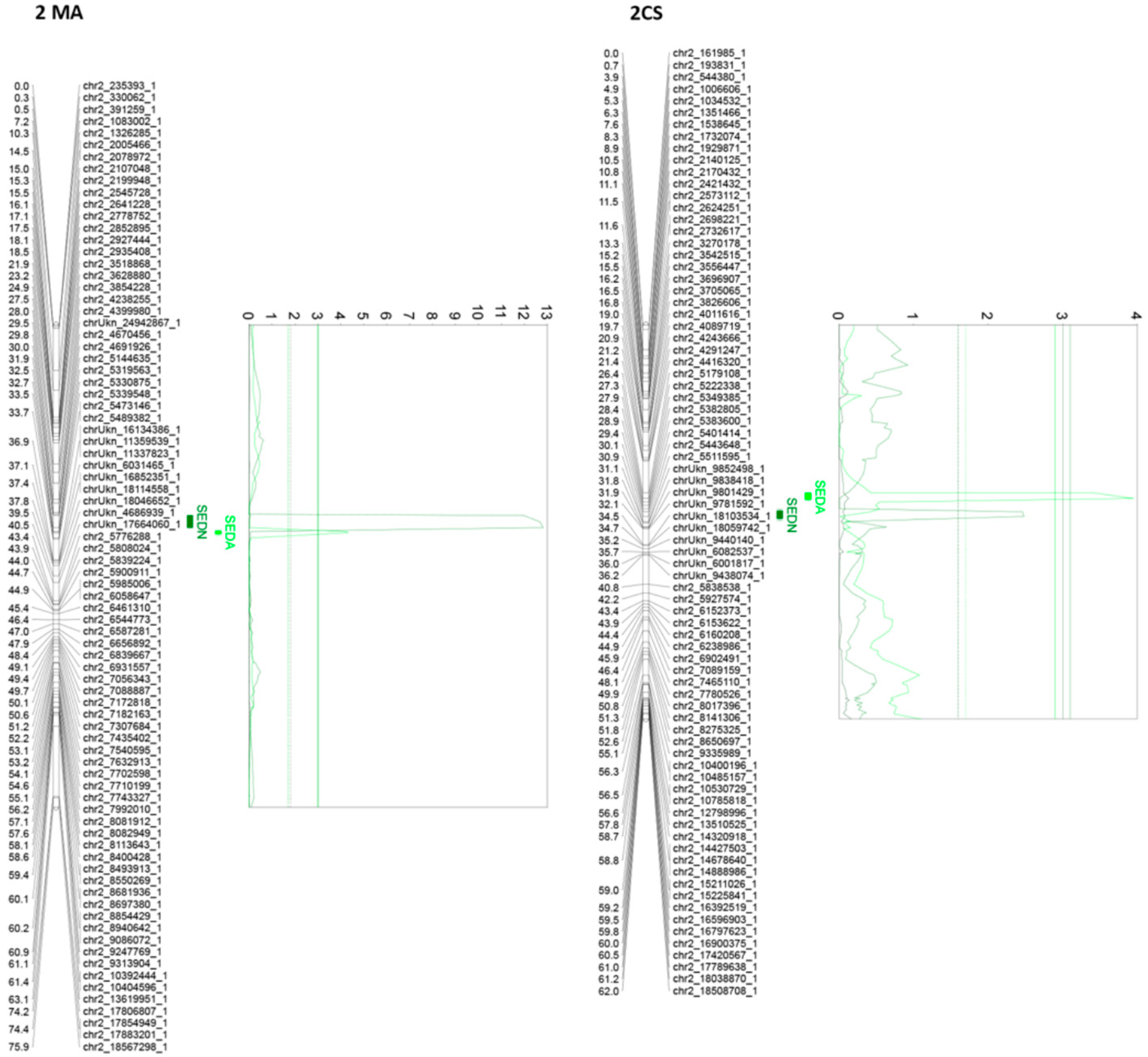
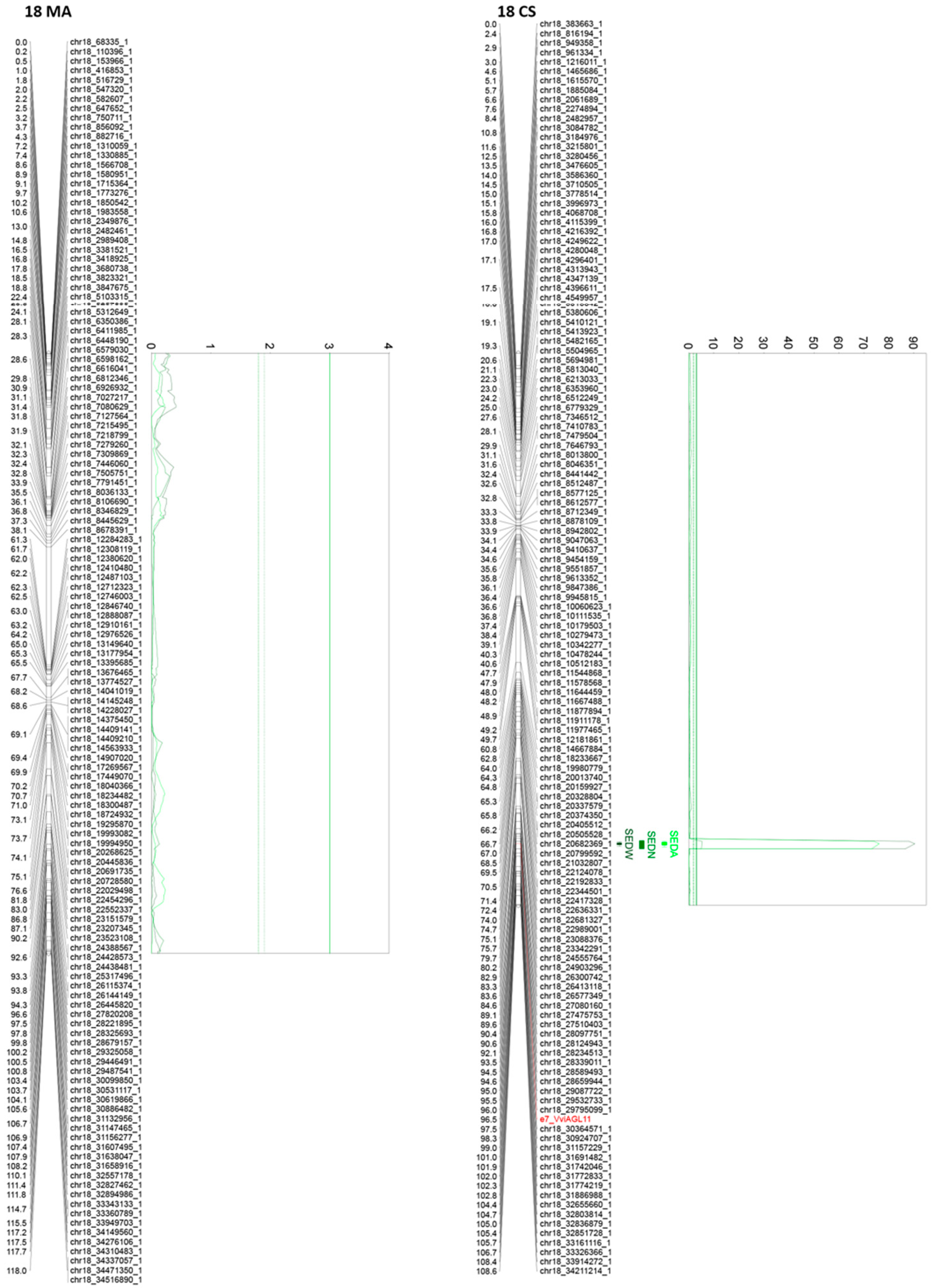
3.3. QTL Analysis
3.4. Multiloci Assisted Selection
3.5. QTL Confidence Intervals and the Identification of Candidate Genes
3.6. Development of Markers for Assisted Selection
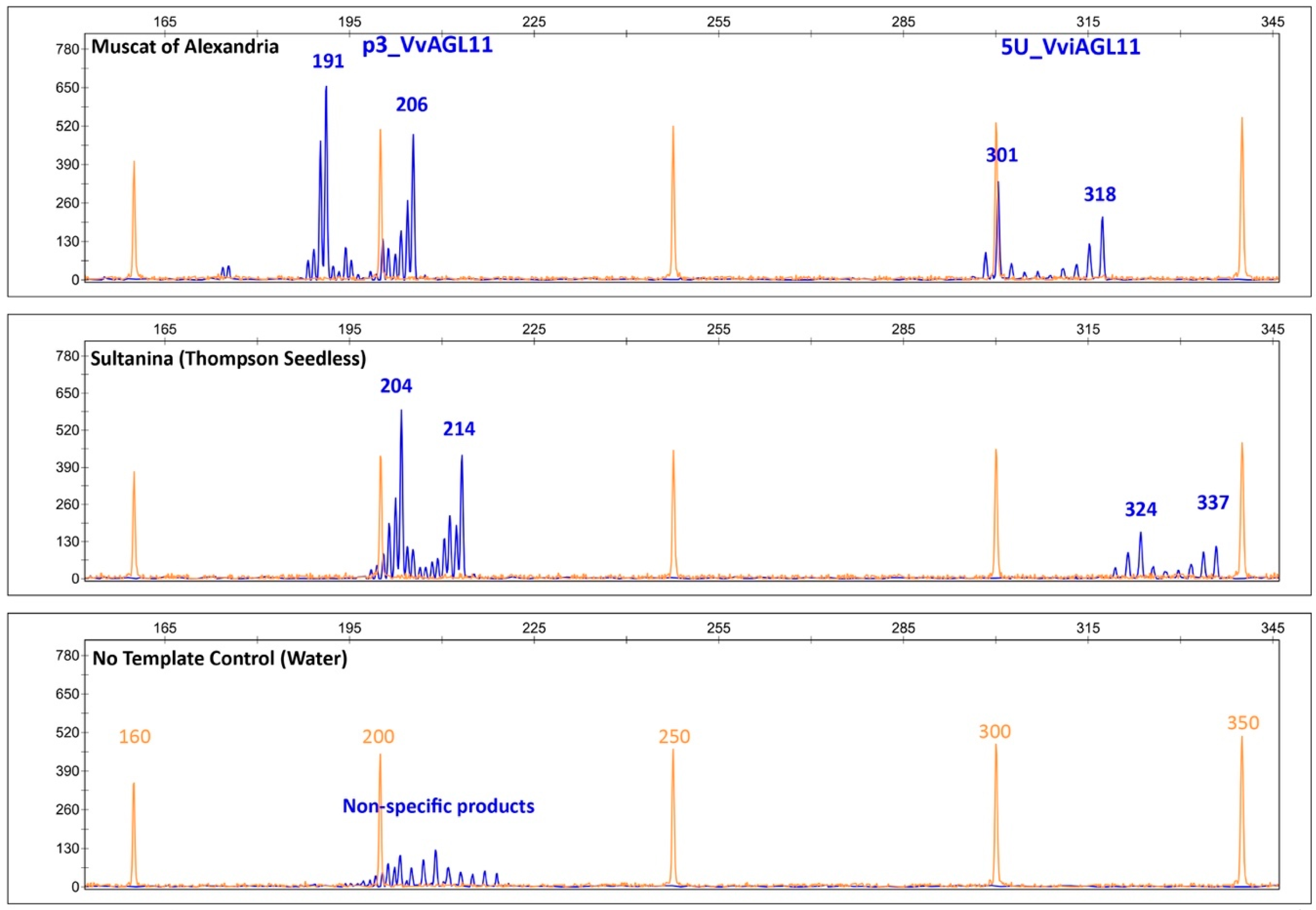
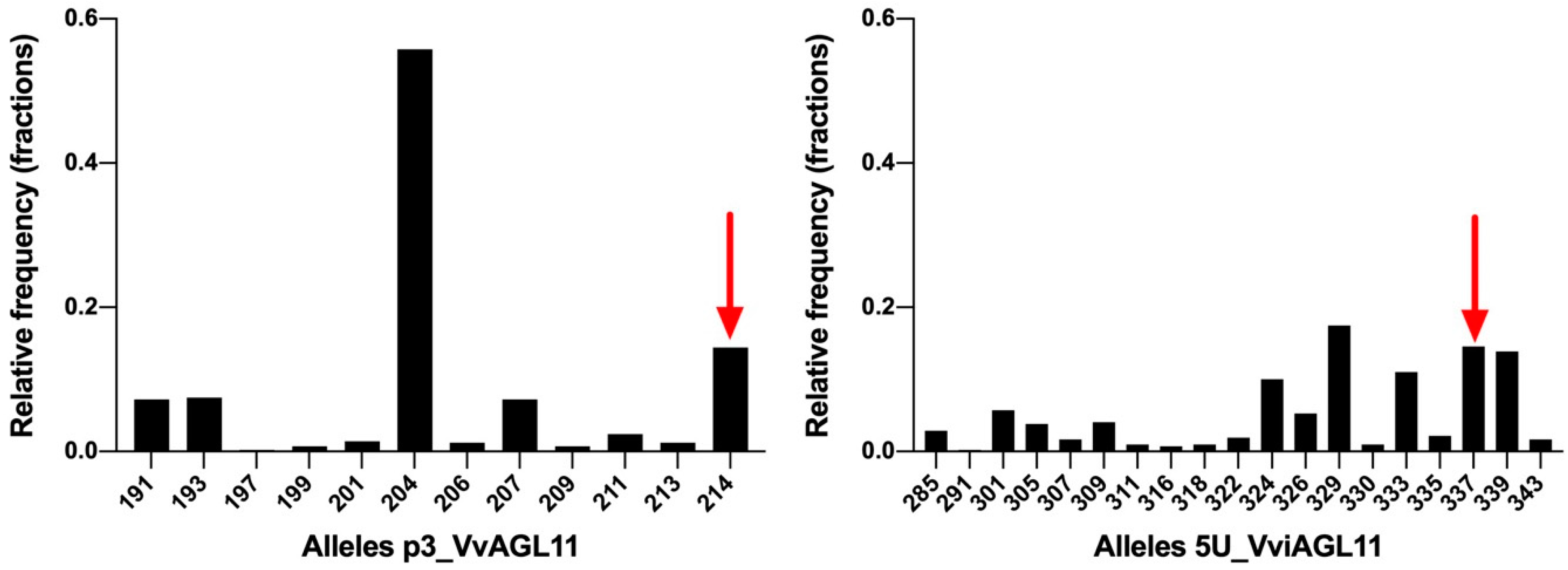
3.6.1. Microsatellite 5U_VviAGL11
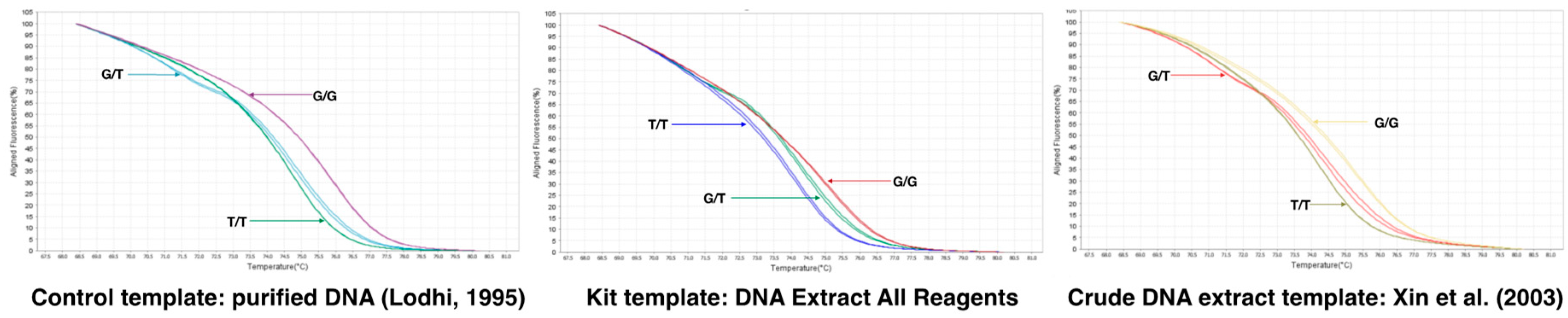
3.6.2. SNP e7_VviAGL11 to Identify the Arg-197-Leu Mutation [G/T] in VviAGL11 by qPCR-HRM
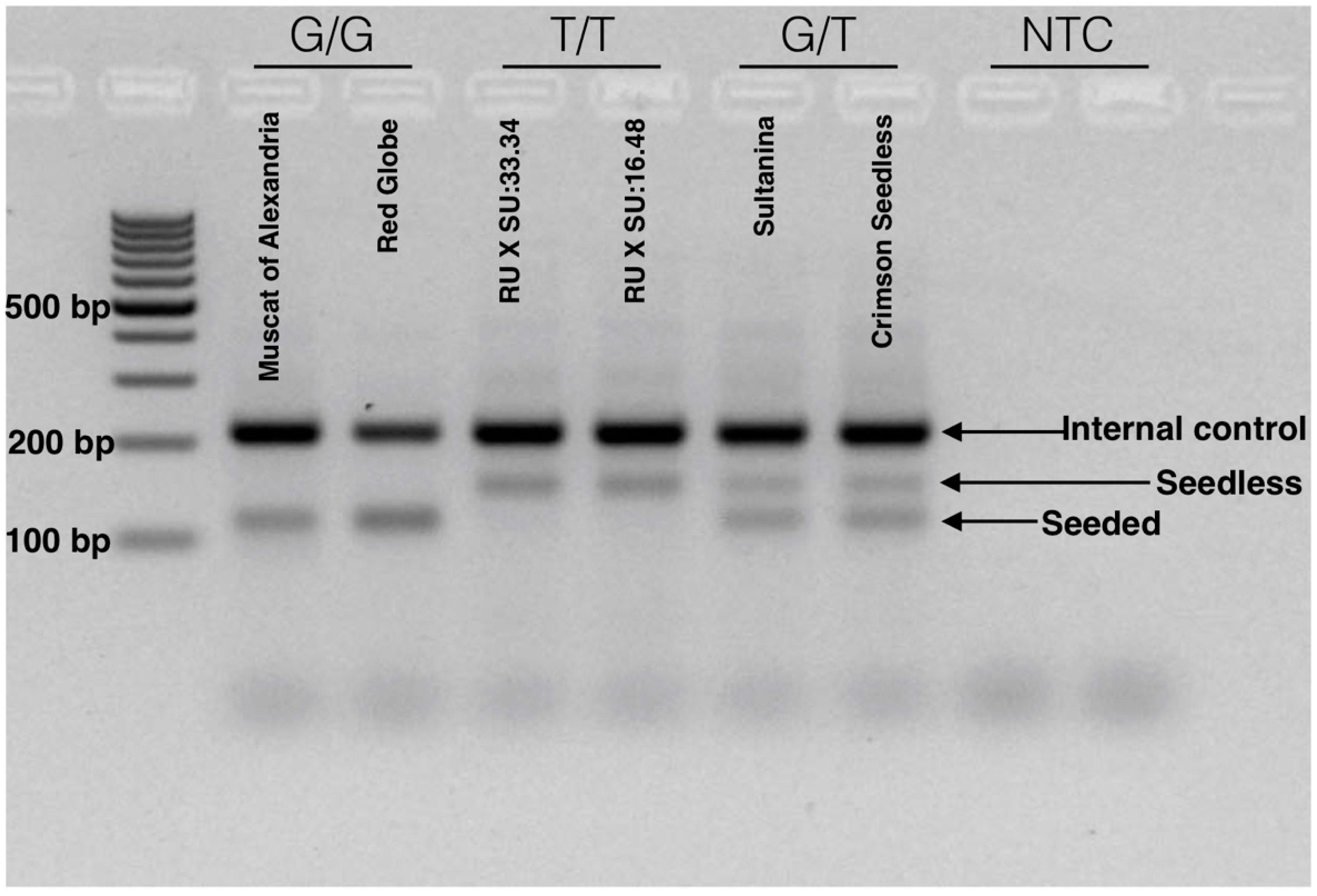
3.6.3. SNP e7_VviAGL11 to Identify the Arg-197-Leu Mutation [G/T] in VviAGL11 by Tetra-Primer ARMS-PCR
3.7. Evaluation of the Performance of SSR 5U_VviAGL11 and SNP e7_VviAGL11 for the Prediction of the Seedless Phenotype in the Context of Breeding
4. Discussions
4.1. Trait Variation
4.2. The Complex Genetic Architecture of Seedlessness
4.3. Candidate Genes
4.4. Multi-QTL Assisted Selection
4.5. Development of Robust, Simple, and Accurate Markers for Assisted Selection of Seedlessness
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Varoquaux, F.; Blanvillain, R.; Delseny, M.; Gallois, P. Less is better: New approaches for seedless fruit production. Trends Biotechnol. 2000, 18, 233–242. [Google Scholar] [CrossRef]
- Zyprian, E.; Eibach, R.; Trapp, O.; Schwander, F.; Toepfer, R. Grapevine breeding under climate change: Applicability of a molecular marker linked to véraison. Vitis J. Grapevine Res. 2018, 57, 119–123. [Google Scholar] [CrossRef]
- Buonassisi, D.; Colombo, M.; Migliaro, D.; Dolzani, C.; Peressotti, E.; Mizzotti, C.; Velasco, R.; Masiero, S.; Perazzolli, M.; Vezzulli, S. Breeding for grapevine downy mildew resistance: A review of “omics” approaches. Euphytica 2017, 213, 103. [Google Scholar] [CrossRef]
- Phillips, J.C.; Rodgriguez, L.P. Beyond Organic: An Overview of Biodynamic Agriculture with Case Examples. In Proceedings of the American Agricultural Economics Association 2006 Annual meeting, Long Beach, CA, USA, 23–26 July 2006. [Google Scholar]
- Seccia, A.; Viscecchia, R.; Nardone, G. Table grapes as functional food: Consumer preferences for health and environmental attributes. BIO Web Conf. 2019, 15. [Google Scholar] [CrossRef]
- Ledbetter, C.A.; Ramming, D.W. Seedlessness in Grapes. Hortic. Rev. 1989, 159–184. [Google Scholar] [CrossRef]
- Lahogue, F.; This, P.; Bouquet, A. Identification of a codominant scar marker linked to the seedlessness character in grapevine. Theor. Appl. Genet. 1998, 97, 950–959. [Google Scholar] [CrossRef]
- Stout, A. Seedlessness in grapes. N.Y. State Agric. Exp. Stn. Tech. Bull. 1936, 1–68. [Google Scholar]
- Mejía, N.; Soto, B.; Guerrero, M.; Casanueva, X.; Houel, C.; Miccono Mde, L.; Ramos, R.; Le Cunff, L.; Boursiquot, J.M.; Hinrichsen, P.; et al. Molecular, genetic and transcriptional evidence for a role of VvAGL11 in stenospermocarpic seedlessness in grapevine. BMC Plant Biol. 2011, 11, 57. [Google Scholar] [CrossRef]
- Bouquet, A.; Danglot, Y. Inheritance of seedlessness in grapevine (Vitis vinifera L.). Vitis 1996, 35, 35–42. [Google Scholar]
- Bergamini, C.; Cardone, M.F.; Anaclerio, A.; Perniola, R.; Pichierri, A.; Genghi, R.; Alba, V.; Forleo, L.R.; Caputo, A.R.; Montemurro, C.; et al. Validation assay of p3_VvAGL11 marker in a wide range of genetic background for early selection of stenospermocarpy in Vitis vinifera L. Mol. Biotechnol. 2013, 54, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Sutton, J.T.; Robertson, B.C.; Jamieson, I.G. Dye shift: A neglected source of genotyping error in molecular ecology. Mol. Ecol. Resour. 2011, 11, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, M.J.; Carpten, J.D.; Smith, J.R. Modulation of non-templated nucleotide addition by Taq DNA polymerase: Primer modifications that facilitate genotyping. Biotechniques 1996, 20, 1004–1006. [Google Scholar] [CrossRef] [PubMed]
- Silkie, S.S.; Tolcher, M.P.; Nelson, K.L. Reagent decontamination to eliminate false-positives in Escherichia coli qPCR. J. Microbiol. Methods 2008, 72, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, M.T.; Warburton, M.L.; Buckler, E.S. Empirical comparison of Simple Sequence Repeats and single nucleotide polymorphisms in assessment of maize diversity and relatedness. PLoS ONE 2007, 2, e1367. [Google Scholar] [CrossRef] [PubMed]
- Royo, C.; Torres-Perez, R.; Mauri, N.; Diestro, N.; Cabezas, J.A.; Marchal, C.; Lacombe, T.; Ibanez, J.; Tornel, M.; Carreno, J.; et al. The Major Origin of Seedless Grapes Is Associated with a Missense Mutation in the MADS-Box Gene VviAGL11. Plant Physiol. 2018, 177, 1234–1253. [Google Scholar] [CrossRef]
- Ocarez, N.; Mejia, N. Suppression of the D-class MADS-box AGL11 gene triggers seedlessness in fleshy fruits. Plant Cell Rep. 2016, 35, 239–254. [Google Scholar] [CrossRef]
- Malabarba, J.; Buffon, V.; Mariath, J.E.A.; Gaeta, M.L.; Dornelas, M.C.; Margis-Pinheiro, M.; Pasquali, G.; Revers, L.F. The MADS-box gene Agamous-like 11 is essential for seed morphogenesis in grapevine. J. Exp. Bot. 2017, 68, 1493–1506. [Google Scholar] [CrossRef]
- Coombe, B.G. Growth Stages of the Grapevine: Adoption of a system for identifying grapevine growth stages. Aust. J. Grape Wine Res. 1995, 1, 104–110. [Google Scholar] [CrossRef]
- Van Ooijen, J.W. MapQTL® 6, Software for the Mapping of Quantitative Trait Loci in Experimental Populations of Diploid Species, 6; Kyazma BV: Wageningen, The Netherlands, 2009. [Google Scholar]
- Di Rienzo, J.A.; Casanoves, F.; Balzarini, M.G.; Gonzalez, L.; Tablada, M.; Robledo, C.W. InfoStat; Grupo InfoStat, FCA, Universidad Nacional de Córdoba: Cordoba, Argentina, 2018. [Google Scholar]
- Doligez, A.; Adam-Blondon, A.F.; Cipriani, G.; Di Gaspero, G.; Laucou, V.; Merdinoglu, D.; Meredith, C.P.; Riaz, S.; Roux, C.; This, P. An integrated SSR map of grapevine based on five mapping populations. Theor. Appl. Genet. 2006, 113, 369–382. [Google Scholar] [CrossRef]
- Thomas, M.R.; A Scott, N.S. Microsatellite repeats in grapevine reveal DNA polymorphisms when analysed as sequence-tagged sites (STSs). Theor. Appl. Genet. 1993, 86, 985–990. [Google Scholar] [CrossRef]
- Houel, C.; Chatbanyong, R.; Doligez, A.; Rienth, M.; Foria, S.; Luchaire, N.; Roux, C.; Adiveze, A.; Lopez, G.; Farnos, M.; et al. Identification of stable QTLs for vegetative and reproductive traits in the microvine (Vitis vinifera L.) using the 18 K Infinium chip. BMC Plant Biol. 2015, 15, 205. [Google Scholar] [CrossRef] [PubMed]
- Jaillon, O.; Aury, J.M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Lodhi, M.; Ye, G.-N.; Weeden, N.; Reisch, B. A simple and efficient method for DNA extraction from grapevine cultivars andVitis species. Plant Mol. Biol. Rep. 1994, 12, 6–13. [Google Scholar] [CrossRef]
- Xin, Z.; Velten, J.P.; Oliver, M.J.; Burke, J.J. High-throughput DNA extraction method suitable for PCR. Biotechniques 2003, 34, 820–824, 826. [Google Scholar] [CrossRef] [PubMed]
- Van Ooijen, J.W. JoinMap®4.1, Software for the Calculation of Genetic Linkage Maps in Experimental Populations, 4.1; Kyazma BV: Wageningen, The Netherlands, 2013. [Google Scholar]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Churchill, G.A.; Doerge, R.W. Empirical threshold values for quantitative trait mapping. Genetics 1994, 138, 963–971. [Google Scholar] [PubMed]
- Doerge, R.W.; Churchill, G.A. Permutation tests for multiple loci affecting a quantitative character. Genetics 1996, 142, 285–294. [Google Scholar]
- Lander, E.S.; Botstein, D. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 1989, 121, 185–199. [Google Scholar]
- Canaguier, A.; Grimplet, J.; Di Gaspero, G.; Scalabrin, S.; Duchene, E.; Choisne, N.; Mohellibi, N.; Guichard, C.; Rombauts, S.; Le Clainche, I.; et al. A new version of the grapevine reference genome assembly (12X.v2) and of its annotation (VCost.v3). Genom. Data 2017, 14, 56–62. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Pluthero, F.G. Rapid purification of high-activity Taq DNA polymerase. Nucleic Acids Res. 1993, 21, 4850–4851. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Dhillon, S.; Ke, X.; Collins, A.R.; Day, I.N. An efficient procedure for genotyping single nucleotide polymorphisms. Nucleic Acids Res. 2001, 29, E88. [Google Scholar] [CrossRef] [PubMed]
- Vihinen, M. How to evaluate performance of prediction methods? Measures and their interpretation in variation effect analysis. BMC Genom. 2012, 13, S2. [Google Scholar] [CrossRef] [PubMed]
- Baldi, P.; Brunak, S.; Chauvin, Y.; Andersen, C.A.; Nielsen, H. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics 2000, 16, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, M.K.; Bowman, J.L.; Sundaresan, V. YABBY polarity genes mediate the repression of KNOX homeobox genes in Arabidopsis. Plant Cell 2002, 14, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Stahle, M.I.; Kuehlich, J.; Staron, L.; von Arnim, A.G.; Golz, J.F. YABBYs and the transcriptional corepressors LEUNIG and LEUNIG_HOMOLOG maintain leaf polarity and meristem activity in Arabidopsis. Plant Cell 2009, 21, 3105–3118. [Google Scholar] [CrossRef]
- Simon, M.K.; Skinner, D.J.; Gallagher, T.L.; Gasser, C.S. Integument Development in Arabidopsis Depends on Interaction of YABBY Protein INNER NO OUTER with Coactivators and Corepressors. Genetics 2017, 207, 1489–1500. [Google Scholar] [CrossRef]
- Keller, M. 2.3. Reproductive Cycle. In The Science of Grapevines: Anatomy and Physiology; Keller, M., Ed.; Academic Press: London, UK, 2010; pp. 68–83. [Google Scholar]
- Ebadi, A.; May, P.; Sedgley, M.; Coombe, B.G. Effect of low temperature near flowering time on ovule development and pollen tube growth in the grapevine (Vitis vinifera L.), cvs Chardonnay and Shiraz. Aust. J. Grape Wine Res. 1995, 1, 11–18. [Google Scholar] [CrossRef]
- Ewart, A.; Kliewer, W.M. Effects of Controlled Day and Night Temperatures and Nitrogen on Fruit-Set, Ovule Fertility, and Fruit Composition of Several Wine Grape Cultivars. Am. J. Enol. Vitic. 1977, 28, 88. [Google Scholar]
- Cheng, C.; Xu, X.; Singer, S.D.; Li, J.; Zhang, H.; Gao, M.; Wang, L.; Song, J.; Wang, X. Effect of GA3 treatment on seed development and seed-related gene expression in grape. PLoS ONE 2013, 8, e80044. [Google Scholar] [CrossRef] [PubMed]
- Doligez, A.; Bouquet, A.; Danglot, Y.; Lahogue, F.; Riaz, S.; Meredith, P.; Edwards, J.; This, P. Genetic mapping of grapevine (Vitis vinifera L.) applied to the detection of QTLs for seedlessness and berry weight. Theor. Appl. Genet. 2002, 105, 780–795. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, J.A.; Cervera, M.T.; Ruiz-Garcia, L.; Carreno, J.; Martinez-Zapater, J.M. A genetic analysis of seed and berry weight in grapevine. Genome 2006, 49, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Costantini, L.; Battilana, J.; Lamaj, F.; Fanizza, G.; Grando, M.S. Berry and phenology-related traits in grapevine (Vitis vinifera L.): From quantitative trait loci to underlying genes. BMC Plant Biol. 2008, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Doligez, A.; Bertrand, Y.; Farnos, M.; Grolier, M.; Romieu, C.; Esnault, F.; Dias, S.; Berger, G.; Francois, P.; Pons, T.; et al. New stable QTLs for berry weight do not colocalize with QTLs for seed traits in cultivated grapevine (Vitis vinifera L.). BMC Plant Biol. 2013, 13, 217. [Google Scholar] [CrossRef] [PubMed]
- Massonnet, M.; Cochetel, N.; Minio, A.; Vondras, A.M.; Muyle, A.; Lin, J.; Garcia, J.F.; Zhou, Y.; Delledonne, M.; Riaz, S.; et al. The genetic basis of sex determination in grapevines (Vitis spp.). bioRxiv 2019. [Google Scholar] [CrossRef]
- Xu, S. Theoretical basis of the Beavis effect. Genetics 2003, 165, 2259–2268. [Google Scholar]
- Malabarba, J.; Buffon, V.; Mariath, J.E.A.; Maraschin, F.S.; Margis-Pinheiro, M.; Pasquali, G.; Revers, L.F. Manipulation of VviAGL11 expression changes the seed content in grapevine (Vitis vinifera L.). Plant Sci. 2018, 269, 126–135. [Google Scholar] [CrossRef]
- Price, A.H. Believe it or not, QTLs are accurate! Trends Plant Sci. 2006, 11, 213–216. [Google Scholar] [CrossRef]
- Sawa, S.; Ito, T.; Shimura, Y.; Okada, K. Filamentous Flower Controls the Formation and Development of Arabidopsis Inflorescences and Floral Meristems. Plant Cell 1999, 11, 69. [Google Scholar] [CrossRef]
- Drews, G.N.; Bowman, J.L.; Meyerowitz, E.M. Negative regulation of the Arabidopsis homeotic gene AGAMOUS by the APETALA2 product. Cell 1991, 65, 991–1002. [Google Scholar] [CrossRef]
- Battilana, J.; Lorenzi, S.; Moreira, F.M.; Moreno-Sanz, P.; Failla, O.; Emanuelli, F.; Grando, M.S. Linkage mapping and molecular diversity at the flower sex locus in wild and cultivated grapevine reveal a prominent SSR haplotype in hermaphrodite plants. Mol. Biotechnol. 2013, 54, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, L.; Sun, X.; Li, Y.; Yao, J.; Nocker, S.V.; Wang, X. Genome-Wide Analysis of the YABBY Gene Family in Grapevine and Functional Characterization of VvYABBY4. Front. Plant Sci. 2019, 10, 1207. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, R.; Gong, P.; Li, S.; Wang, Y.; Zhang, C. Gene Cloning, Expression and Enzyme Activity of Vitis vinifera Vacuolar Processing Enzymes (VvVPEs). PLoS ONE 2016, 11, e0160945. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Wei, R.; Li, Y.; Wang, R.; Tang, Y.; Wang, L.; Zhu, H.; Wang, Y.; Zhang, C. Molecular cloning and functional characterization of a seed-specific VvβVPE gene promoter from Vitis vinifera. Planta 2019, 250, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Li, Y.; Tang, Y.; Wei, R.; Huijun, Z.; Wang, Y.; Zhang, C. Vacuolar processing enzyme (VvβVPE) from Vitis vinifera, processes seed proteins during ovule development, and accelerates seed germination in VvβVPE heterologously over-expressed Arabidopsis. Plant Sci. 2018, 274, 420–431. [Google Scholar] [CrossRef]
- Nakaune, S.; Yamada, K.; Kondo, M.; Kato, T.; Tabata, S.; Nishimura, M.; Hara-Nishimura, I. A vacuolar processing enzyme, deltaVPE, is involved in seed coat formation at the early stage of seed development. Plant Cell 2005, 17, 876–887. [Google Scholar] [CrossRef]
- Minio, A.; Massonnet, M.; Figueroa-Balderas, R.; Vondras, A.M.; Blanco-Ulate, B.; Cantu, D. Iso-Seq Allows Genome-Independent Transcriptome Profiling of Grape Berry Development. G3 Genes Gen. Genet. 2019, 9, 755. [Google Scholar] [CrossRef]
- Minio, A.; Lin, J.; Gaut, B.S.; Cantu, D. How Single Molecule Real-Time Sequencing and Haplotype Phasing Have Enabled Reference-Grade Diploid Genome Assembly of Wine Grapes. Front. Plant Sci. 2017, 8, 826. [Google Scholar] [CrossRef]
- Wang, R.; Dobritsa, A.A. Exine and Aperture Patterns on the Pollen Surface: Their Formation and Roles in Plant Reproduction. Ann. Plant Rev. Online 2018, 589–628. [Google Scholar] [CrossRef]
- Dobritsa, A.A.; Kirkpatrick, A.B.; Reeder, S.H.; Li, P.; Owen, H.A. Pollen Aperture Factor INP1 Acts Late in Aperture Formation by Excluding Specific Membrane Domains from Exine Deposition. Plant Physiol. 2018, 176, 326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, Y.; Lin, X.; Hong, X.; Zhu, Y.; Li, W.; He, W.; An, F.; Guo, H. Adenine Phosphoribosyl Transferase 1 is a Key Enzyme Catalyzing Cytokinin Conversion from Nucleobases to Nucleotides in Arabidopsis. Mol Plant 2013, 6, 1661–1672. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, M.; Manrique, S.; Guazzotti, A.; Quadrelli, N.E.; Mendes, M.A.; Benkova, E.; Colombo, L. Cytokinin response factors integrate auxin and cytokinin pathways for female reproductive organ development. Development 2016, 143, 4419. [Google Scholar] [CrossRef] [PubMed]
- Nesi, N.; Jond, C.; Debeaujon, I.; Caboche, M.; Lepiniec, L. The Arabidopsis TT2; Gene Encodes an R2R3 MYB Domain Protein That Acts as a Key Determinant for Proanthocyanidin Accumulation in Developing Seed. Plant Cell 2001, 13, 2099. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, W.; Shi, J.; Xu, J.; Zhang, D. MYB56 Encoding a R2R3 MYB Transcription Factor Regulates Seed Size in Arabidopsis thaliana. J. Integr. Plant Biol. 2013, 55, 1166–1178. [Google Scholar] [CrossRef]
- Li, D.; Jin, C.; Duan, S.; Zhu, Y.; Qi, S.; Liu, K.; Gao, C.; Ma, H.; Zhang, M.; Liao, Y.; et al. MYB89 Transcription Factor Represses Seed Oil Accumulation. Plant Physiol. 2017, 173, 1211–1225. [Google Scholar] [CrossRef]
- Kim, J.H.; Hyun, W.Y.; Nguyen, H.N.; Jeong, C.Y.; Xiong, L.; Hong, S.-W.; Lee, H. AtMyb7, a subgroup 4 R2R3 Myb, negatively regulates ABA-induced inhibition of seed germination by blocking the expression of the bZIP transcription factor ABI5. Plant Cell Environ. 2015, 38, 559–571. [Google Scholar] [CrossRef]
- Kobayashi, K.; Suzuki, T.; Iwata, E.; Nakamichi, N.; Suzuki, T.; Chen, P.; Ohtani, M.; Ishida, T.; Hosoya, H.; Müller, S.; et al. Transcriptional repression by MYB3R proteins regulates plant organ growth. EMBO J. 2015, 34, 1992–2007. [Google Scholar] [CrossRef]
- Schwinn, K.E.; Ngo, H.; Kenel, F.; Brummell, D.A.; Albert, N.W.; McCallum, J.A.; Pither-Joyce, M.; Crowhurst, R.N.; Eady, C.; Davies, K.M. The Onion (Allium cepa L.) R2R3-MYB Gene MYB1 Regulates Anthocyanin Biosynthesis. Front. Plant Sci. 2016, 7, 1865. [Google Scholar] [CrossRef]
- Shin, R.; Burch, A.Y.; Huppert, K.A.; Tiwari, S.B.; Murphy, A.S.; Guilfoyle, T.J.; Schachtman, D.P. The Arabidopsis Transcription Factor MYB77 Modulates Auxin Signal Transduction. Plant Cell 2007, 19, 2440. [Google Scholar] [CrossRef]
- Ishida, T.; Hattori, S.; Sano, R.; Inoue, K.; Shirano, Y.; Hayashi, H.; Shibata, D.; Sato, S.; Kato, T.; Tabata, S.; et al. Arabidopsis TRANSPARENT TESTA GLABRA2 Is Directly Regulated by R2R3 MYB Transcription Factors and Is Involved in Regulation of GLABRA2 Transcription in Epidermal Differentiation. Plant Cell 2007, 19, 2531. [Google Scholar] [CrossRef] [PubMed]
- Doughty, J.; Aljabri, M.; Scott, R.J. Flavonoids and the regulation of seed size in Arabidopsis. Biochem. Soc. Trans. 2014, 42, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Debeaujon, I.; Peeters, A.J.M.; Léon-Kloosterziel, K.M.; Koornneef, M. The TRANSPARENT TESTA12; Gene of Arabidopsis Encodes a Multidrug Secondary Transporter-like Protein Required for Flavonoid Sequestration in Vacuoles of the Seed Coat Endothelium. Plant Cell 2001, 13, 853. [Google Scholar] [CrossRef] [PubMed]
- Kondou, Y.; Nakazawa, M.; Kawashima, M.; Ichikawa, T.; Yoshizumi, T.; Suzuki, K.; Ishikawa, A.; Koshi, T.; Matsui, R.; Muto, S.; et al. RETARDED GROWTH OF EMBRYO1, a new basic helix-loop-helix protein, expresses in endosperm to control embryo growth. Plant Physiol. 2008, 147, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, P.; Lecourieux, D.; Gomes, E.; Delrot, S.; Lecourieux, F. The grape berry-specific basic helix-loop-helix transcription factor VvCEB1 affects cell size. J. Exp. Bot. 2013, 64, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.D.; Yim, W.C.; Liu, D.; Hu, R.; Yang, X.; Cushman, J.C. A Vitis vinifera basic helix–loop–helix transcription factor enhances plant cell size, vegetative biomass and reproductive yield. Plant Biotechnol. J. 2018, 16, 1595–1615. [Google Scholar] [CrossRef]
- Jain, P.; Shah, K.; Sharma, N.; Kaur, R.; Singh, J.; Vinson, C.; Rishi, V. A-ZIP53, a dominant negative reveals the molecular mechanism of heterodimerization between bZIP53, bZIP10 and bZIP25 involved in Arabidopsis seed maturation. Sci. Rep. 2017, 7, 14343. [Google Scholar] [CrossRef]
- Zinsmeister, J.; Lalanne, D.; Terrasson, E.; Chatelain, E.; Vandecasteele, C.; Vu, B.L.; Dubois-Laurent, C.; Geoffriau, E.; Signor, C.L.; Dalmais, M.; et al. ABI5 Is a Regulator of Seed Maturation and Longevity in Legumes. Plant Cell 2016, 28, 2735–2754. [Google Scholar] [CrossRef]
- Alonso, R.; Oñate-Sánchez, L.; Weltmeier, F.; Ehlert, A.; Diaz, I.; Dietrich, K.; Vicente-Carbajosa, J.; Dröge-Laser, W. A pivotal role of the basic leucine zipper transcription factor bZIP53 in the regulation of Arabidopsis seed maturation gene expression based on heterodimerization and protein complex formation. Plant Cell 2009, 21, 1747–1761. [Google Scholar] [CrossRef]
- Nambara, E.; McCourt, P.; Naito, S. A regulatory role for the ABI3 gene in the establishment of embryo maturation in Arabidopsis. Development 1995, 121. [Google Scholar]
- Shu, K.; Zhang, H.; Wang, S.; Chen, M.; Wu, Y.; Tang, S.; Liu, C.; Feng, Y.; Cao, X.; Xie, Q. ABI4 regulates primary seed dormancy by regulating the biogenesis of abscisic acid and gibberellins in arabidopsis. PLoS Genet. 2013, 9, e1003577. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-H.; Kim, B.; Song, S.-K.; Heo, J.-O.; Yu, N.-I.; Lee, S.A.; Kim, M.; Kim, D.G.; Sohn, S.O.; Lim, C.E.; et al. Large-scale analysis of the GRAS gene family in Arabidopsis thaliana. Plant Mol. Biol. 2008, 67, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Haecker, A.; Groß-Hardt, R.; Geiges, B.; Sarkar, A.; Breuninger, H.; Herrmann, M.; Laux, T. Expression dynamics of WOX genes mark cell fate decisions during early embryonic patterning in Arabidopsis thaliana. Development 2004, 131, 657. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Mitsuda, N.; Ohme-Takagi, M. Arabidopsis WUSCHEL is a bifunctional transcription factor that acts as a repressor in stem cell regulation and as an activator in floral patterning. Plant Cell 2009, 21, 3493–3505. [Google Scholar] [CrossRef]
- Park, S.O.; Zheng, Z.; Oppenheimer, D.G.; Hauser, B.A. The PRETTY FEW SEEDS2 gene encodes an Arabidopsis homeodomain protein that regulates ovule development. Development 2005, 132, 841. [Google Scholar] [CrossRef]
- Dai, M.; Hu, Y.; Zhao, Y.; Liu, H.; Zhou, D.X. A WUSCHEL-LIKE HOMEOBOX gene represses a YABBY gene expression required for rice leaf development. Plant Physiol. 2007, 144, 380–390. [Google Scholar] [CrossRef]
- Krizek, B.A.; Prost, V.; Macias, A. AINTEGUMENTA Promotes Petal Identity and Acts as a Negative Regulator of AGAMOUS. Plant Cell 2000, 12, 1357. [Google Scholar] [CrossRef]
- Elliott, R.C.; Betzner, A.S.; Huttner, E.; Oakes, M.P.; Tucker, W.Q.; Gerentes, D.; Perez, P.; Smyth, D.R. AINTEGUMENTA, an APETALA2-like gene of Arabidopsis with pleiotropic roles in ovule development and floral organ growth. Plant Cell 1996, 8, 155. [Google Scholar] [CrossRef]
- Bowman, J.L.; Smyth, D.R.; Meyerowitz, E.M. Genes directing flower development in Arabidopsis. Plant Cell 1989, 1, 37. [Google Scholar] [CrossRef]
- Jack, T.; Fox, G.L.; Meyerowitz, E.M. Arabidopsis homeotic gene APETALA3 ectopic expression: Transcriptional and posttranscriptional regulation determine floral organ identity. Cell 1994, 76, 703–716. [Google Scholar] [CrossRef]
- de Martino, G.; Pan, I.; Emmanuel, E.; Levy, A.; Irish, V.F. Functional Analyses of Two Tomato APETALA3 Genes Demonstrate Diversification in Their Roles in Regulating Floral Development. Plant Cell 2006, 18, 1833. [Google Scholar] [CrossRef] [PubMed]
- Strelin, M.M.; Aizen, M.A. The interplay between ovule number, pollination and resources as determinants of seed set in a modular plant. PeerJ 2018, 6, e5384. [Google Scholar] [CrossRef] [PubMed]
- Toepfer, R.; Hausmann, L.; Harst, M.; Maul, E.; Zyprian, E.; Eibach, R. New Horizons for Grapevine Breeding; Federal Research Centre for Cultivated Plants, Institute for Grapevine Breeding Geilweilerhof: Siebeldingen, Germany, 2011; Volume 5, pp. 79–100. [Google Scholar]
- Youn Young, H.; Chan Jin, J.; Jung-Ho, N.; Sung-Min, J.; Jong-Chul, N.; Kyung Ho, M.; and Kyo-Sun, P. Analysis of Genetic Relationship of Seedless Germplasm and Validation Assay of the P3_VvAGL11 Marker Linked to Seedlessness in Grapevines. Korean J. Breed. Sci. 2014, 46, 28–36. [Google Scholar] [CrossRef]
- Karastan, O.; Mulyukina, N.; Papina, O.; Plachinda, G. ПОЛІМОРФІЗМ ІНТРАГЕННОГО МІКРОСАТЕЛІТНОГО ЛОКУСУ P3_VVAGL11, ЗЧЕПЛЕНОГО З ОЗНАКОЮ БЕЗНАСІННЄВОСТІ У ВИНОГРАДУ (VITIS VINIFERA L.). Вісник Львівського університету. Серія біологічна 2015, 70, 90–99. [Google Scholar]
- Conner, P.; Gunawan, G.; Clark, J. Characterization of the p3-VvAGL11 Marker for Stenospermocarpic Seedlessness in Euvitis × Muscadinia Grape Hybrid Progenies. J. Am. Soc. Hortic. Sci. 2018, 143, 167–172. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Chrom. | Position (cM) | SNP Cofactor | LOD | % Expl. Var | Mean Genotype a | Mean Genotype b | LOD | Stable | QTL | Support | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2015 | 2016 | 2017 | 2018 | |||||||||||
| SEDW | 9MA | 33.108 | chr9_5511355_1 | 2.26 | 1.7 | 0.0867 | 0.0753 | 0.98 | 0.93 | 0.72 | 3.28 | No | Significant | BLUP + 1 Season |
| 13MA | 1.345 | chr13_810814_1 | 1.96 | 1.5 | 0.0740 | 0.0870 | 1.99 | 0.89 | 1.54 | 1.93 | Yes | Suggestive | BLUP + 2 Seasons | |
| 13MA | 61.553 | chr13_14817494_1 | 2.39 | 1.8 | 0.0742 | 0.0886 | 0.53 | 1.5 | 2.5 | 1.88 | Yes | Suggestive | BLUP + 2 Seasons | |
| 4CS | 63.423 | chr4_19816331_1 | 2.33 | 0.9 | 0.0885 | 0.0740 | 2.24 | 1.03 | 1.78 | 1.47 | Yes | Suggestive | BLUP + 2 Seasons | |
| 14CS | 10.51 | chr14_3367810_1 | 2.06 | 0.8 | 0.0758 | 0.0880 | 0.19 | 1.02 | 0.93 | 1.49 | No | Suggestive | BLUP only | |
| 18CS | 96.474 | e7_VviAGL11 | 90.4 | 48.4 | 0.1052 | 0.0410 | 38.85 | 73.61 | 58.93 | 46.78 | Yes | Significant | BLUP + 4 seasons | |
| SEDN | 1MA | 23.448 | chr1_4720746_1 | 4.03 | 2.8 | 2.8760 | 3.2148 | 1.95 | 2.85 | 0.88 | 1.91 | Yes | Significant | BLUP + 3 seasons |
| 2MA | 31.857 | chr2_5144635_1 | 12.81 | 9.2 | 2.6574 | 3.3845 | 5.41 | 6.59 | 0.73 | 16.5 | Yes | Significant | BLUP + 3 seasons | |
| 8MA | 53.979 | chr8_16567159_1 | 2.06 | 1.4 | 3.2588 | 2.9028 | 1.5 | 0.74 | 0.89 | 0.64 | No | Suggestive | BLUP only | |
| 13MA | 1.345 | chr13_810814_1 | 2.49 | 1.7 | 2.8896 | 3.1877 | 1.19 | 0.74 | 0.24 | 2.03 | No | Suggestive | BLUP + 1 Season | |
| 1CS | 68.878 | chr1_20882467_1 | 2.82 | 1.9 | 3.2313 | 2.8568 | 0.24 | 4.49 | 0.59 | 0.85 | No | Suggestive | BLUP + 1 Season | |
| 2CS | 30.109 | chr2_5443648_1 | 2.48 | 1.6 | 3.2588 | 2.9172 | 1.61 | 0.36 | 1.3 | 3.7 | No | Suggestive | BLUP + 1 Season | |
| 7CS | 99.657 | chr7_24375532_1 | 2.46 | 1.6 | 3.1993 | 2.8989 | 0.79 | 0.98 | 0.13 | 2.97 | No | Suggestive | BLUP + 1 Season | |
| 12CS | 4.672 | chr12_1741069_1 | 2.95 | 1.9 | 3.2576 | 2.8558 | 0.94 | 3.43 | 2.35 | 0.43 | Yes | Suggestive | BLUP + 2 Seasons | |
| 12CS | 65.968 | chr12_22397742_1 | 2.76 | 1.8 | 2.8865 | 3.2123 | 2.02 | 1.43 | 0.35 | 1.73 | Yes | Suggestive | BLUP + 2 Seasons | |
| 18CS | 96.474 | e7_VviAGL11 | 5.27 | 3.5 | 3.2574 | 2.7010 | 3.51 | 1.87 | 0.61 | 4.84 | Yes | Significant | BLUP + 2 Seasons | |
| SEDA | 2MA | 32.691 | chr2_5330875_1 | 4.32 | 3.2 | 0.0797 | 0.0968 | 0.25 | 3.57 | 0.18 | 8.96 | Yes | Significant | BLUP + 2 Seasons |
| 9MA | 8.439 | chr9_1387529_1 | 2.56 | 1.9 | 0.0815 | 0.0944 | 2.28 | 1.10 | 0.72 | 2.24 | Yes | Suggestive | BLUP + 2 Seasons | |
| 13MA | 61.22 | chr13_14790761_1 | 2.27 | 1.7 | 0.0938 | 0.0826 | 0.13 | 2.15 | 5.15 | 1.06 | Yes | Suggestive | BLUP + 2 Seasons | |
| 2CS | 27.274 | chr2_5222338_1 | 3.95 | 1.6 | 0.0788 | 0.0933 | 0.74 | 0.09 | 1.43 | 4.42 | No | Significant | BLUP + 1 Season | |
| 8CS | 50.684 | chr8_17020635_1 | 2.66 | 1.1 | 0.0878 | 0.0972 | 0.02 | 1.79 | 1.92 | 2.52 | Yes | Suggestive | BLUP + 3 seasons | |
| 10CS | 54.671 | chr10_8168185_1 | 3.15 | 1.3 | 0.0827 | 0.0827 | 1.16 | 0.92 | 0.59 | 2.89 | No | Significant | BLUP + 1 Season | |
| 14CS | 10.51 | chr14_3367810_1 | 1.93 | 0.8 | 0.0878 | 0.0834 | 0.33 | 0.48 | 1.22 | 0.89 | No | Suggestive | BLUP only | |
| 18CS | 96.474 | e7_VviAGL11 | 76.08 | 42.5 | 0.1107 | 0.0506 | 20.74 | 43.37 | 53.98 | 38.99 | Yes | Significant | BLUP + 4 seasons | |
| Multiloci Assisted Selection in Bi-Parental Population, One-Way ANOVA Model Summary for SEDW, n = 571 | ||||||||||||
| Order by % Var. Expl. | Parental | QTL Cofactor | Individual Favorable Genotype | Nº of QTLs | Factors | Cumulative Favorable Genotype (Ideotype) | Ideotype N | Ideotype Mean (g) | Ideotype Std. Dev. | Ideotype 95% C.I. | R² % (Adjusted) | p-Value |
| 1 | CS | e7_VviAGL11 | b | 1 | 2 | b | 217 | 0.0412 | 0.0256 | (0.03776;0.04462) | 52.11% | 0.000 |
| 2 | MA | chr13_14817494_1 | a | 2 | 4 | ba | 122 | 0.0363 | 0.0233 | (0.03208;0.04045) | 53.71% | 0.000 |
| 3 | MA | chr9_5511355_1 | b | 3 | 8 | bab | 70 | 0.0321 | 0.0184 | (0.02773;0.03653) | 54.38% | 0.000 |
| 4 | MA | chr13_810814_1 | a | 4 | 16 | baba | 43 | 0.0316 | 0.0213 | (0.02505;0.03815) | 55.41% | 0.000 |
| 5 | CS | chr4_19816331_1 | b | 5 | 32 | babab | 29 | 0.0308 | 0.0234 | (0.02187;0.03968) | 55.98% | 0.000 |
| Multiloci Assisted Selection in Bi-Parental Population, One-Way ANOVA Model Summary for SEDN, n = 572 | ||||||||||||
| Order by % Var. Expl. | Parental | QTL Cofactor | Individual Favorable Genotype | Nº of QTLs | Factors | Cumulative Favorable Genotype (Ideotype) | Ideotype N | Ideotype Mean (number) | Ideotype Std. Dev. | Ideotype 95% C.I. | R² % (Adjusted) | p-Value |
| 1 | MA | chr2_5144635_1 | a | 1 | 2 | a | 267 | 2.6574 | 1.1862 | (2.5145;2.8003) | 9.15% | 0.000 |
| 2 | CS | e7_VviAGL11 | b | 2 | 4 | ab | 100 | 2.4220 | 1.0830 | (2.208;2.637) | 14.63% | 0.000 |
| 3 | MA | chr1_4720746_1 | a | 3 | 8 | aba | 44 | 2.1980 | 1.0930 | (1.866;2.530) | 17.09% | 0.000 |
| 4 | CS | chr1_20882467_1 | b | 4 | 16 | abab | 19 | 1.8610 | 0.9310 | (1.412;2.310) | 19.46% | 0.000 |
| 5 | CS | chr12_1741069_1 | b | 5 | 32 | ababb | 13 | 1.7950 | 0.8360 | (1.289;2.300) | 21.72% | 0.000 |
| Multiloci Assisted Selection in Bi-Parental Population, One-Way ANOVA Model Summary for SEDA, n = 569 | ||||||||||||
| Order by % Var. Expl. | Parental | QTL Cofactor | Individual Favorable Genotype | Nº of QTLs | Factors | Cumulative Favorable Genotype (Ideotype) | Ideotype N | Ideotype Mean (g) | Ideotype Std. Dev. | Ideotype 95% C.I. | R² % (Adjusted) | p-Value |
| 1 | CS | e7_VviAGL11 | b | 1 | 2 | b | 218 | 0.05056 | 0.02813 | (0.04681;0.05432) | 43.87% | 0.000 |
| 2 | MA | chr2_5330875_1 | a | 2 | 4 | ba | 119 | 0.05004 | 0.02631 | (0.04526;0.05481) | 48.83% | 0.000 |
| 3 | MA | chr9_1387529_1 | a | 3 | 8 | baa | 71 | 0.04831 | 0.02505 | (0.04238;0.05424) | 49.06% | 0.000 |
| 4 | MA | chr13_14790761_1 | b | 4 | 16 | baab | 34 | 0.03942 | 0.01989 | (0.03248;0.04636) | 49.82% | 0.000 |
| 5 | CS | chr2_5222338_1 | a | 5 | 32 | baaba | 15 | 0.03227 | 0.01562 | (0.02362;0.04092) | 51.92% | 0.000 |
| Type of Material | Detail of Material | Number of Genotypes | p3_VvAGL11 SSR | U_VviAGL11 SSR | e7_VviAGL11 SNP |
|---|---|---|---|---|---|
| Variety collection | Seeded Varieties | 161 | 151 Seeded and 10 Seedless | 151 Seeded and 10 Seedless | 161 Seeded [G/G] |
| Variety collection | Seedless Varieties | 48 | 48 Seedless | 48 Seedless | 48 Seedless [G/T] |
| Experimental genotype | Control homozygous seedless | 2 | 2 Seedless | 2 Seedless | 2 Seedless [T/T] |
| F1 progeny | Seeded Progeny (RG × AR) | 31 | 30 Seeded and 1 Seedless | 30 Seeded and 1 Seedless | 30 Seeded [G/G] and 1 Seedless [G/T] |
| F1 progeny | Seedless Progeny (RG × AR) | 17 | 4 Seeded and 13 Seedless | 4 Seeded and 13 Seedless | 4 Seeded [G/G] and 13 Seedless [G/T] |
| F1 progeny | Seeded (RG × RS) | 18 | 16 Seeded, 1 Seedless and 1 NA | 17 Seeded and 1 Seedless | 17 Seeded [G/G] and 1 Seedless [G/T] |
| F1 progeny | Seedless (RG × RS) | 30 | 1 Seeded and 29 Seedless | 1 Seeded and 29 Seedless | 1 Seeded [G/G] and 29 Seedless [G/T] |
| Breeding material | Seeded Progeny (multiple crosses) | 56 | 54 Seeded and 2 Seedless | 54 Seeded and 2 Seedless | 54 Seeded [G/G] and 2 Seedless [G/T] |
| Breeding material | Seedless Progeny (multiple crosses) | 118 | 110 Seedless and 8 NA | 118 Seedless | 118 Seedless [G/T] |
| Other Vitis species | Other Vitis | 9 | 9 Seeded | 9 Seeded | 8 Seeded [G/G] |
| Other Vitaceae members | Other Vitaceae | 4 | 4 Seeded | 4 NA | 5 Seeded [G/G] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ocarez, N.; Jiménez, N.; Núñez, R.; Perniola, R.; Marsico, A.D.; Cardone, M.F.; Bergamini, C.; Mejía, N. Unraveling the Deep Genetic Architecture for Seedlessness in Grapevine and the Development and Validation of a New Set of Markers for VviAGL11-Based Gene-Assisted Selection. Genes 2020, 11, 151. https://doi.org/10.3390/genes11020151
Ocarez N, Jiménez N, Núñez R, Perniola R, Marsico AD, Cardone MF, Bergamini C, Mejía N. Unraveling the Deep Genetic Architecture for Seedlessness in Grapevine and the Development and Validation of a New Set of Markers for VviAGL11-Based Gene-Assisted Selection. Genes. 2020; 11(2):151. https://doi.org/10.3390/genes11020151
Chicago/Turabian StyleOcarez, Nallatt, Nicolás Jiménez, Reynaldo Núñez, Rocco Perniola, Antonio Domenico Marsico, Maria Francesca Cardone, Carlo Bergamini, and Nilo Mejía. 2020. "Unraveling the Deep Genetic Architecture for Seedlessness in Grapevine and the Development and Validation of a New Set of Markers for VviAGL11-Based Gene-Assisted Selection" Genes 11, no. 2: 151. https://doi.org/10.3390/genes11020151
APA StyleOcarez, N., Jiménez, N., Núñez, R., Perniola, R., Marsico, A. D., Cardone, M. F., Bergamini, C., & Mejía, N. (2020). Unraveling the Deep Genetic Architecture for Seedlessness in Grapevine and the Development and Validation of a New Set of Markers for VviAGL11-Based Gene-Assisted Selection. Genes, 11(2), 151. https://doi.org/10.3390/genes11020151