You Had Me at “MAGIC”!: Four Barley MAGIC Populations Reveal Novel Resistance QTL for Powdery Mildew

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
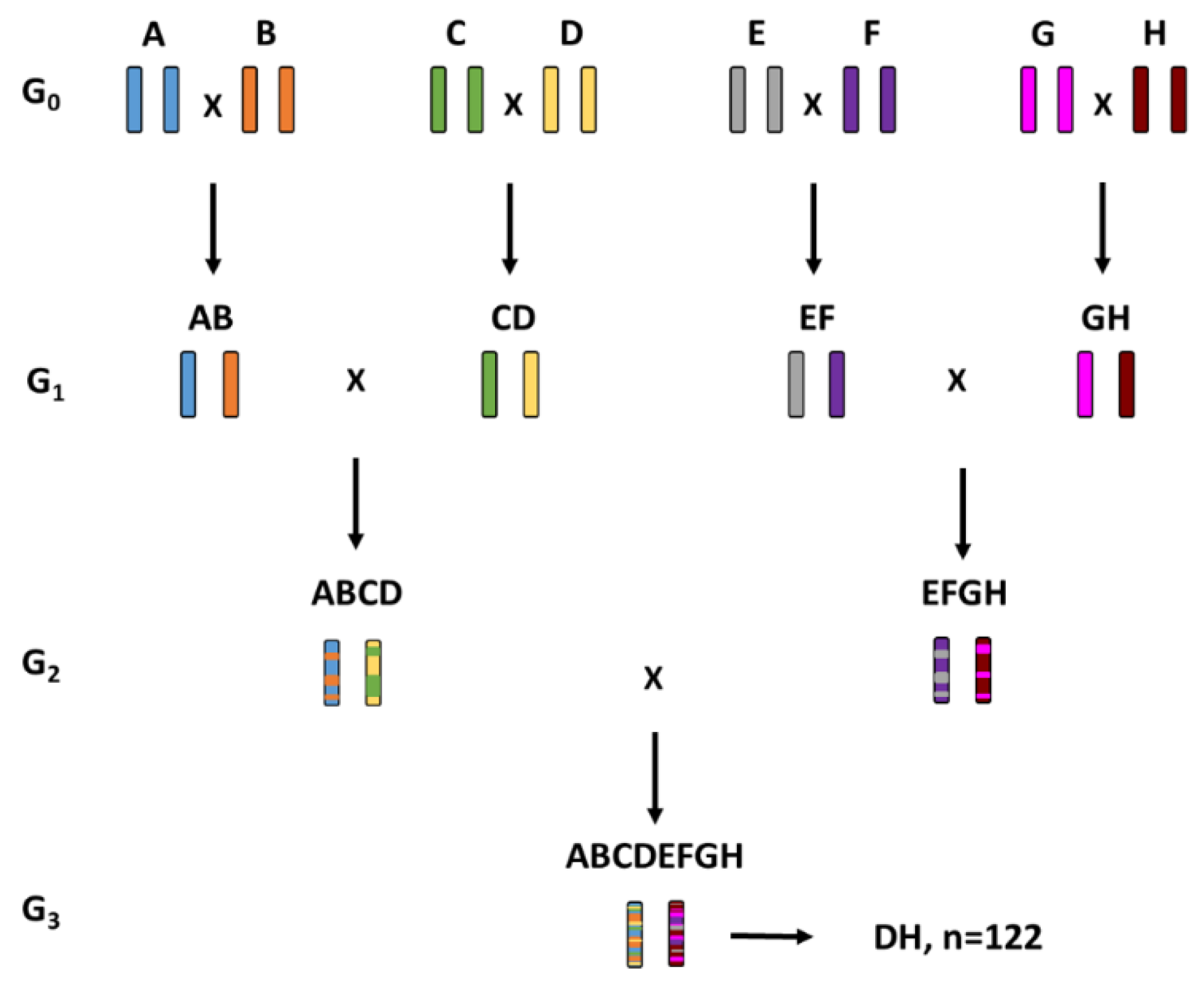
2.1. Multi-Parent Advanced Generation Inter-Cross (MAGIC) Populations
2.2. Field Trials and Phenotypic Evaluation
2.3. Statistical Analysis
2.4. Genotyping and SNP Filtering
2.5. Population Structure and LINKAGE Disequilibrium
2.6. Association Mapping
2.7. Candidate Gene Indentification and Haplotype Formation
3. Results
3.1. Panel Evaluation
3.2. Population Structure and Linkage Disequilibrium
3.3. Model Selection
3.4. Marker Trait Associations (MTAs)
3.5. Candidate Genes for the Identified QTL
3.6. Allele Combinations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Langridge, P. Economic and Academic Importance of Barley. In The Barley Genome; Stein, N., Muehlbauer, G.J., Eds.; Springer International Publishing: Cham, Germany, 2018; pp. 1–10. ISBN 978-3-319-92528-8. [Google Scholar]
- Hilmarsson, H.S.; Göransson, M.; Lillemo, M.; Kristjánsdóttir, Þ.A.; Hermannsson, J.; Hallsson, J.H. An overview of barley breeding and variety trials in Iceland in 1987–2014. Icel. Agric. Sci. 2017, 30, 13–28. [Google Scholar] [CrossRef]
- Glawe, D.A. The powdery mildews: A review of the world’s most familiar (yet poorly known) plant pathogens. Annu. Rev. Phytopathol. 2008, 46, 27–51. [Google Scholar] [CrossRef] [PubMed]
- Czembor, J.H. Resistance to powdery mildew in selections from Moroccan barley landraces. Euphytica 2002, 125, 397–409. [Google Scholar] [CrossRef]
- Schüller, C.; Backes, G.; Fischbeck, G.; Jahoor, A. RFLP markers to identify the alleles on the Mla locus conferring powdery mildew resistance in barley. Theor. Appl. Genet. 1992, 84, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Gobelman-Werner, K.; Morroll, S.M.; Kurth, J.; Mao, L.; Wing, R.; Leister, D.; Schulze-Lefert, P.; Wise, R.P. The Mla (powdery mildew) resistance cluster is associated with three NBS-LRR gene families and suppressed recombination within a 240-kb DNA interval on chromosome 5S (1HS) of barley. Genetics 1999, 153, 1929–1948. [Google Scholar] [PubMed]
- Jørgensen, J.H.; Wolfe, M. Genetics of powdery mildew resistance in barley. Crit. Rev. Plant Sci. 1994, 13, 97–119. [Google Scholar] [CrossRef]
- Giese, H.; Holm-Jensen, A.G.; Jensen, H.P.; Jensen, J. Localization of the Laevigatum powdery mildew resistance gene to barley chromosome 2 by the use of RFLP markers. Theor. Appl. Genet. 1993, 85, 897–900. [Google Scholar] [CrossRef]
- Piechota, U.; Czembor, P.C.; Słowacki, P.; Czembor, J.H. Identifying a novel powdery mildew resistance gene in a barley landrace from Morocco. J. Appl. Genet. 2019, 60, 243–254. [Google Scholar] [CrossRef]
- Görg, R.; Hollricher, K.; Schulze-Lefert, P. Functional analysis and RFLP-mediated mapping of the Mlg resistance locus in barley. Plant J. 1993, 3, 857–866. [Google Scholar] [CrossRef]
- Schönfeld, M.; Ragni, A.; Fischbeck, G.; Jahoor, A. RFLP mapping of three new loci for resistance genes to powdery mildew (Erysiphe graminis f. sp. hordei) in barley. Theor. Appl. Genet. 1996, 93, 48–56. [Google Scholar] [CrossRef]
- Dreiseitl, A. Specific resistance of barley to powdery mildew, its use and beyond. A concise critical review. Genes 2020, 11, 971. [Google Scholar] [CrossRef] [PubMed]
- Mejlhede, N.; Kyjovska, Z.; Backes, G.; Burhenne, K.; Rasmussen, S.K.; Jahoor, A. EcoTILLING for the identification of allelic variation in the powdery mildew resistance genes mlo and Mla of barley. Plant Breed. 2006, 125, 461–467. [Google Scholar] [CrossRef]
- Seeholzer, S.; Tsuchimatsu, T.; Jordan, T.; Bieri, S.; Pajonk, S.; Yang, W.; Jahoor, A.; Shimizu, K.K.; Keller, B.; Schulze-Lefert, P. Diversity at the Mla powdery mildew resistance locus from cultivated barley reveals sites of positive selection. Mol. Plant Microbe Interact. 2010, 23, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Giese, H.; Jørgensen, J.H.; Jensen, H.P.; Jensen, J. Linkage relationships of ten powdery mildew resistance genes on barley chromosome 5. Hereditas 1981, 95, 43–50. [Google Scholar] [CrossRef]
- Giese, H. Powdery mildew resistance genes in the Ml-a and Ml-k regions on barley chromosome 5. Hereditas 1981, 95, 51–62. [Google Scholar] [CrossRef]
- Kintzios, S.; Jahoor, A.; Fischbeck, G. Powdery-mildew-resistance genes Mla29 and Mla32 in H. spontaneum derived winter-barley ines. Plant Breed. 1995, 266, 265–267. [Google Scholar] [CrossRef]
- Jahoor, A.; Fischbeck, G. Identification of New Genes for Mildew Resistance of Barley at the Mia Locus in Lines Derived from Hordeum spontaneum. Plant Breed. 1993, 110, 116–122. [Google Scholar] [CrossRef]
- Wei, F.; Wing, R.A.; Wise, R.P. Genome dynamics and evolution of the Mla (powdery mildew) resistance locus in barley. Plant Cell 2002, 14, 1903–1917. [Google Scholar] [CrossRef]
- Halterman, D.; Zhou, F.; Wei, F.; Wise, R.P.; Schulze-lefert, P. The MLA6 coiled-coil, NBS-LRR protein confers AvrMla6—Dependent resistance specificity to Blumeria graminis f. sp. hordei in barley and wheat. Plant J. 2001, 25, 335–348. [Google Scholar] [CrossRef]
- Halterman, D.A.; Wei, F.; Wise, R.P. Powdery mildew-induced Mla mRNAs are alternatively spliced and contain multiple upstream open reading frames. Plant Physiol. 2003, 131, 558–567. [Google Scholar] [CrossRef]
- Shen, Q.H.; Zhou, F.; Bieri, S.; Haizel, T.; Shirasu, K.; Schulze-Lefert, P. Recognition specificity and RAR1/SGT1 dependence in barley Mla disease resistance genes to the powdery mildew fungus. Plant Cell 2003, 15, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Kurth, J.; Wei, F.; Elliott, C.; Valè, G.; Yahiaoui, N.; Keller, B.; Somerville, S.; Wise, R.; Schulze-lefert, P. Cell-Autonomous Expression of Barley Mla1 Confers Race-Specific Resistance to the Powdery Mildew Fungus via a Rar1—Independent Signaling Pathway. Plant Cell 2001, 13, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Halterman, D.A.; Wise, R.P. A single-amino acid substitution in the sixth leucine-rich repeat of barley MLA6 and MLA13 alleviates dependence on RAR1 for disease resistance signaling. Plant J. 2004, 38, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Büschges, R.; Hollricher, K.; Panstruga, R.; Simons, G.; Wolter, M.; Frijters, A.; Van Daelen, R.; Van der Lee, T.; Diergaarde, P.; Groenendijk, J.; et al. The barley Mlo gene: A novel control element of plant pathogen resistance. Cell 1997, 88, 695–705. [Google Scholar] [CrossRef]
- Freisleben, R.; Lein, A. Über die Auffindung einer mehltauresistenten Mutante nach Röntgenbestrahlung einer anfälligen reinen Linie von Sommergerste. Naturwissenschaften 1942, 30, 608. [Google Scholar] [CrossRef]
- Jørgensen, J.H. Discovery, characterization and exploitation of Mlo powdery mildew resistance in barley. Euphytica 1992, 63, 141–152. [Google Scholar] [CrossRef]
- Reinstädler, A.; Müller, J.; Czembor, J.H.; Piffanelli, P.; Panstruga, R. Novel induced mlo mutant alleles in combination with site-directed mutagenesis reveal functionally important domains in the heptahelical barley Mlo protein. BMC Plant Biol. 2010, 10, 31. [Google Scholar] [CrossRef]
- Freialdenhoven, A.; Peterhansel, C.; Kurth, J.; Kreuzaler, F.; Schulze-Lefert, P. ldentification of Genes Required for the Function of Non-Race-Specific. Plant Cell 1996, 8, 5–14. [Google Scholar] [CrossRef]
- Devoto, A.; Piffanelli, P.; Nilsson, I.M.; Wallin, E.; Panstruga, R.; Von Heijne, G.; Schulze-Lefert, P. Topology, subcellular localization, and sequence diversity of the Mlo family in plants. J. Biol. Chem. 1999, 274, 34993–35004. [Google Scholar] [CrossRef]
- Devoto, A.; Hartmann, H.A.; Piffanelli, P.; Elliott, C.; Simmons, C.; Taramino, G.; Goh, C.S.; Cohen, F.E.; Emerson, B.C.; Schulze-Lefert, P.; et al. Molecular phylogeny and evolution of the plant-specific seven-transmembrane MLO family. J. Mol. Evol. 2003, 56, 77–88. [Google Scholar] [CrossRef]
- Kusch, S.; Panstruga, R. Mlo-based resistance: An apparently universal “weapon” to defeat powdery mildew disease. Mol. Plant Microbe Interact. 2017, 30, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Jarosch, B.; Kogel, K.H.; Schaffrath, U. The ambivalence of the barley Mlo locus: Mutations conferring resistance against powdery mildew (Blumeria graminis f. sp. hordei) Enhance susceptibility to the rice blast fungus Magnaporthe grisea. Mol. Plant Microbe Interact. 1999, 12, 508–514. [Google Scholar] [CrossRef]
- Kumar, J.; Hückelhoven, R.; Beckhove, U.; Nagarajan, S.; Kogel, K.H. A compromised Mlo pathway affects the response of barley to the necrotrophic fungus Bipolaris sorokiniana (teleomorph: Cochliobolus sativus) and its toxins. Phytopathology 2001, 91, 127–133. [Google Scholar] [CrossRef] [PubMed]
- McGrann, G.R.D.; Stavrinides, A.; Russell, J.R.; Corbitt, M.M.; Booth, A.; Chartrain, L.; Thomas, W.T.B.; Brown, J.K.M. A trade off between mlo resistance to powdery mildew and increased susceptibility of barley. J. Exp. Bot. 2014, 65, 1025–1037. [Google Scholar] [CrossRef]
- Havis, N.D.; Brown, J.K.M.; Clemente, G.; Frei, P.; Jedryczka, M.; Kaczmarek, J.; Kaczmarek, M.; Matusinsky, P.; McGrann, G.R.D.; Pereyra, S.; et al. Ramularia collo-cygni—An emerging pathogen of barley crops. Phytopathology 2015, 105, 895–904. [Google Scholar] [CrossRef]
- Alqudah, A.M.; Sallam, A.; Stephen Baenziger, P.; Börner, A. GWAS: Fast-forwarding gene identification and characterization in temperate Cereals: Lessons from Barley—A review. J. Adv. Res. 2020, 22, 119–135. [Google Scholar] [CrossRef]
- Rafalski, J.A. Association genetics in crop improvement. Curr. Opin. Plant Biol. 2010, 13, 174–180. [Google Scholar] [CrossRef]
- Cavanagh, C.R.; Morell, M.; Mackay, I.; Powell, W. From mutations to MAGIC: Resources for gene discovery, validation and delivery in crop plants. Curr. Opin. Plant Biol. 2008, 11, 215–221. [Google Scholar] [CrossRef]
- Huang, X.; Han, B. Natural Variations and Genome-Wide Association Studies in Crop Plants. Annu. Rev. Plant Biol. 2014, 65, 531–551. [Google Scholar] [CrossRef]
- Huang, B.E.; Verbyla, K.L.; Verbyla, A.P.; Raghavan, C.; Singh, V.K.; Gaur, P.; Leung, H.; Varshney, R.K.; Cavanagh, C.R. MAGIC populations in crops: Current status and future prospects. Theor. Appl. Genet. 2015, 128, 999–1017. [Google Scholar] [CrossRef]
- Scott, M.F.; Ladejobi, O.; Amer, S.; Bentley, A.R.; Biernaskie, J.; Boden, S.A.; Clark, M.; Dell’Acqua, M.; Dixon, L.E.; Filippi, C.V.; et al. Multi-parent populations in crops: A toolbox integrating genomics and genetic mapping with breeding. Heredity 2020, 125, 396–416. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Thyssen, G.N.; Jenkins, J.N.; Zeng, L.; Delhom, C.D.; McCarty, J.C.; Deng, D.D.; Hinchliffe, D.J.; Jones, D.C.; Fang, D.D. A MAGIC population-based genome-wide association study reveals functional association of GhRBB1_A07 gene with superior fiber quality in cotton. BMC Genom. 2016, 17, 903. [Google Scholar] [CrossRef] [PubMed]
- Ongom, P.O.; Ejeta, G. Mating design and genetic structure of a multi-parent advanced generation intercross (MAGIC) population of sorghum (Sorghum bicolor (L.) moench). G3 Genes Genomes Genet. 2018, 8, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zhao, H.; Yu, K.; Wang, T.; Khattak, A.N.; Tian, E. Development of a multiparent advanced generation intercross (MAGIC) population for genetic exploitation of complex traits in Brassica juncea: Glucosinolate content as an example. Plant Breed. 2020, 319, 779–789. [Google Scholar] [CrossRef]
- Bandillo, N.; Raghavan, C.; Muyco, P.A.; Sevilla, M.A.L.; Lobina, I.T.; Dilla-Ermita, C.J.; Tung, C.W.; McCouch, S.; Thomson, M.; Mauleon, R.; et al. Multi-parent advanced generation inter-cross (MAGIC) populations in rice: Progress and potential for genetics research and breeding. Rice 2013, 6, 1–15. [Google Scholar] [CrossRef]
- Meng, L.; Wang, B.; Zhao, X.; Ponce, K.; Qian, Q.; Ye, G. Association mapping of ferrous, zinc, and aluminum tolerance at the seedling stage in Indica rice using MAGIC populations. Front. Plant Sci. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Meng, L.; Zhao, X.; Ponce, K.; Ye, G.; Leung, H. QTL mapping for agronomic traits using multi-parent advanced generation inter-cross (MAGIC) populations derived from diverse elite indica rice lines. Field Crop. Res. 2016, 189, 19–42. [Google Scholar] [CrossRef]
- Ogawa, D.; Nonoue, Y.; Tsunematsu, H.; Kanno, N.; Yamamoto, T.; Yonemaru, J.I. Discovery of QTL alleles for grain shape in the Japan-MAGIC rice population using haplotype information. G3 Genes Genomes Genet. 2018, 8, 3559–3565. [Google Scholar] [CrossRef]
- Huang, B.E.; George, A.W.; Forrest, K.L.; Kilian, A.; Hayden, M.J.; Morell, M.K.; Cavanagh, C.R. A multiparent advanced generation inter-cross population for genetic analysis in wheat. Plant Biotechnol. J. 2012, 10, 826–839. [Google Scholar] [CrossRef]
- Sannemann, W.; Lisker, A.; Maurer, A.; Léon, J.; Kazman, E.; Cöster, H.; Holzapfel, J.; Kempf, H.; Korzun, V.; Ebmeyer, E.; et al. Adaptive selection of founder segments and epistatic control of plant height in the MAGIC winter wheat population WM-800. BMC Genom. 2018, 19, 1–16. [Google Scholar] [CrossRef]
- Stadlmeier, M.; Hartl, L.; Mohler, V. Usefulness of a multiparent advanced generation intercross population with a greatly reduced mating design for genetic studies in winter wheat. Front. Plant Sci. 2018, 871, 1–12. [Google Scholar] [CrossRef]
- Mackay, I.J.; Bansept-Basler, P.; Bentley, A.R.; Cockram, J.; Gosman, N.; Greenland, A.J.; Horsnell, R.; Howells, R.; O’Sullivan, D.M.; Rose, G.A.; et al. An eight-parent multiparent advanced generation inter-cross population for winter-sown wheat: Creation, properties, and validation. G3 Genes Genomes Genet. 2014, 4, 1603–1610. [Google Scholar] [CrossRef]
- Dell’Acqua, M.; Gatti, D.M.; Pea, G.; Cattonaro, F.; Coppens, F.; Magris, G.; Hlaing, A.L.; Aung, H.H.; Nelissen, H.; Baute, J.; et al. Genetic properties of the MAGIC maize population: A new platform for high definition QTL mapping in Zea mays. Genome Biol. 2015, 16, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Sannemann, W.; Huang, B.E.; Mathew, B.; Léon, J. Multi-parent advanced generation inter-cross in barley: High-resolution quantitative trait locus mapping for flowering time as a proof of concept. Mol. Breed. 2015, 35, 86. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Jia, Z.; Dunwell, J.M. Editorial: The applications of new multi-locus gwas methodologies in the genetic dissection of complex traits. Front. Plant Sci. 2019, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Czembor, P.C.; Czembor, J.H. DNA polymorphisms among near-isogenic lines for powdery-mildew-resistant genes in barley detected by primers targeting resistance-gene analogs. Can. J. Plant Pathol. 2002, 24, 499–503. [Google Scholar] [CrossRef]
- Silvar, C.; Dhif, H.; Igartua, E.; Kopahnke, D.; Gracia, M.P.; Lasa, J.M.; Ordon, F.; Casas, A.M. Identification of quantitative trait loci for resistance to powdery mildew in a Spanish barley landrace. Mol. Breed. 2010, 25, 581–592. [Google Scholar] [CrossRef]
- Ge, X.; Deng, W.; Lee, Z.Z.; Lopez-Ruiz, F.J.; Schweizer, P.; Ellwood, S.R. Tempered mlo broad-spectrum resistance to barley powdery mildew in an Ethiopian landrace. Sci. Rep. 2016, 6, 29558. [Google Scholar] [CrossRef]
- Abdel-Ghani, A.H.; Al-Ameiri, N.S.; Karajeh, M.R. Resistance of barley landraces and wild barley populations to powdery mildew in Jordan. Phytopathol. Mediterr. 2008, 47, 92–97. [Google Scholar] [CrossRef]
- Řepková, J.; Dreiseitl, A.; Lízal, P.; Kyjovská, Z.; Teturová, K.; Psotková, R.; Jahoor, A. Identification of resistance genes against powdery mildew in four accessions of Hordeum vulgare ssp. spontaneum. Euphytica 2006, 151, 23–30. [Google Scholar] [CrossRef]
- Bengtsson, T.; Åhman, I.; Manninen, O.; Reitan, L.; Christerson, T.; Due Jensen, J.; Krusell, L.; Jahoor, A.; Orabi, J.; Jensen, J.D.; et al. A novel QTL for powdery mildew resistance in nordic spring barley (Hordeum vulgare L. ssp. vulgare) revealed by genome-wide association study. Front. Plant Sci. 2017, 8, 1954. [Google Scholar] [CrossRef] [PubMed]
- Revelle, W. Psych: Procedures for Personality and Psychological Research; Northwestern University: Evanston, IL, USA, 2014; Volume 165, pp. 1–10. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Bates, D.; Mächler, M.; Bolker, B.M.; Walker, S.C. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67. [Google Scholar] [CrossRef]
- Pinheiro, J.; Bates, D.; DebRoy, S.; Sarkar, D.; EISPACK authors; Heisterkamp, S.; Van Willigen, B.; R Core Team. nlme: Linear and Nonlinear Mixed Effects Models; R Package Version 3; 2020; pp. 1–149. Available online: https://cran.r-project.org/package=nlme (accessed on 29 May 2020).
- Bayer, M.M.; Rapazote-Flores, P.; Ganal, M.; Hedley, P.E.; Macaulay, M.; Plieske, J.; Ramsay, L.; Russell, J.R.; Shaw, P.D.; Thomas, W.T.B.; et al. Development and evaluation of a barley 50k iSelect SNP array. Front. Plant Sci. 2017, 8, 1792. [Google Scholar] [CrossRef] [PubMed]
- Åhman, I.; Bengtsson, T. Introgression of resistance to Rhopalosiphum padi L. from wild barley into cultivated barley facilitated by doubled haploid and molecular marker techniques. Theor. Appl. Genet. 2019, 132, 1397–1408. [Google Scholar] [CrossRef]
- Mascher, M.; Gundlach, H.; Himmelbach, A.; Beier, S.; Twardziok, S.O.; Wicker, T.; Radchuk, V.; Dockter, C.; Hedley, P.E.; Russell, J.R.; et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 2017, 544, 427–433. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Boudiar, R.; Casas, A.M.; Igartua, E.; Contreras-Moreira, B. BARLEYMAP: Physical and genetic mapping of nucleotide sequences and annotation of surrounding loci in barley. Mol. Breed. 2015, 35, 1–11. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Covarrubias-Pazaran, G. Genome-Assisted Prediction of Quantitative Traits Using the R Package sommer. PLoS ONE 2016, 11, e0156744. [Google Scholar] [CrossRef]
- Zhang, Z.; Ersoz, E.; Lai, C.Q.; Todhunter, R.J.; Tiwari, H.K.; Gore, M.A.; Bradbury, P.J.; Yu, J.; Arnett, D.K.; Ordovas, J.M.; et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 2010, 42, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Segura, V.; Vilhjálmsson, B.J.; Platt, A.; Korte, A.; Seren, Ü.; Long, Q.; Nordborg, M. An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 2012, 44, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef]
- Van Raden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Li, M.X.; Yeung, J.M.Y.; Cherny, S.S.; Sham, P.C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 2012, 131, 747–756. [Google Scholar] [CrossRef]
- Andersen, S.B.; Due, I.K.; Olesen, A. The Response of Anther Culture in a Genetically Wide Material of Winter Wheat (Triticum aestivum L.). Plant Breed. 1987, 99, 181–186. [Google Scholar] [CrossRef]
- Milner, S.G.; Jost, M.; Taketa, S.; Mazón, E.R.; Himmelbach, A.; Oppermann, M.; Weise, S.; Knüpffer, H.; Basterrechea, M.; König, P.; et al. Genebank genomics highlights the diversity of a global barley collection. Nat. Genet. 2019, 51, 319–326. [Google Scholar] [CrossRef]
- Nordborg, M.; Borevitz, J.O.; Bergelson, J.; Berry, C.C.; Chory, J.; Hagenblad, J.; Kreitman, M.; Maloof, J.N.; Noyes, T.; Oefner, P.J.; et al. The extent of linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 2002, 30, 190–193. [Google Scholar] [CrossRef]
- Flint-Garcia, S.A.; Thornsberry, J.M.; Edward IV, S.B. Structure of Linkage Disequilibrium in Plants. Annu. Rev. Plant Biol. 2003, 54, 357–374. [Google Scholar] [CrossRef]
- Hinze, K.; Thompson, R.D.; Ritter, E.; Salamini, F.; Schulze-lefert, P. Restriction fragment length polymorphism-mediated targeting of the ml-o resistance locus in barley (Hordeum vulgare). Proc. Natl. Acad. Sci. USA 1991, 88, 3691–3695. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.L.; Liu, S.; Hall, M.D.; Brooks, W.S.; Chao, S.; Muehlbauer, G.J.; Baik, B.K.; Steffenson, B.; Griffey, C.A. Marker-trait associations in Virginia Tech winter barley identified using genome-wide mapping. Theor. Appl. Genet. 2013, 126, 693–710. [Google Scholar] [CrossRef] [PubMed]
- Caldo, R.A.; Nettleton, D.; Wise, R.P. Interaction-dependent gene expression in Mla-specified response to barley powdery mildew. Plant Cell 2004, 16, 2514–2528. [Google Scholar] [CrossRef] [PubMed]
- Soldánová, M.; Ištvánek, J.; Řepková, J.; Dreiseitl, A. Newly discovered genes for resistance to powdery mildew in the subtelomeric region of the short arm of barley chromosome 7H. Czech J. Genet. Plant Breed. 2013, 49, 95–102. [Google Scholar] [CrossRef]
- Yun, S.J.; Gyenis, L.; Hayes, P.M.; Matus, I.; Smith, K.P.; Steffenson, B.J.; Muehlbauer, G.J. Quantitative trait loci for multiple disease resistance in wild barley. Crop Sci. 2005, 45, 2563–2572. [Google Scholar] [CrossRef]
- Aghnoum, R.; Marcel, T.C.; Johrde, A.; Pecchioni, N.; Schweizer, P.; Niks, R.E. Basal host resistance of barley to powdery mildew: Connecting quantitative trait loci and candidate genes. Mol. Plant Microbe Interact. 2010, 23, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Von Korff, M.; Wang, H.; Léon, J.; Pillen, K. AB-QTL analysis in spring barley. I. Detection of resistance genes against powdery mildew, leaf rust and scald introgressed from wild barley. Theor. Appl. Genet. 2005, 111, 583–590. [Google Scholar] [CrossRef]
- Schmalenbach, I.; Körber, N.; Pillen, K. Selecting a set of wild barley introgression lines and verification of QTL effects for resistance to powdery mildew and leaf rust. Theor. Appl. Genet. 2008, 117, 1093–1106. [Google Scholar] [CrossRef]
- Panstruga, R.; Parker, J.E.; Schulze-lefert, P. SnapShot: Plant Immune Response Pathways. Cell 2009, 136, 6–8. [Google Scholar] [CrossRef]
- Kumar, M.; Brar, A.; Yadav, M.; Chawade, A.; Vivekanand, V.; Pareek, N. Chitinases—Potential Candidates for Enhanced Plant Resistance towards Fungal Pathogens. Agriculture 2018, 8, 88. [Google Scholar] [CrossRef]
- Pandey, S.P.; Somssich, I.E. The role of WRKY transcription factors in plant immunity. Plant Physiol. 2009, 150, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, J.; Schober, M.S.; Shirley, N.J.; Singh, R.R.; Jacobs, A.K.; Douchkov, D.; Schweizer, P.; Fincher, G.B.; Burton, R.A.; Little, A. Down-regulation of the glucan synthase-like 6 gene (HvGsl6) in barley leads to decreased callose accumulation and increased cell wall penetration by Blumeria graminis f. sp. hordei. New Phytol. 2016, 212, 434–443. [Google Scholar] [CrossRef] [PubMed]
- González, A.M.; Marcel, T.C.; Kohutova, Z.; Stam, P.; van der Linden, C.G.; Niks, R.E. Peroxidase profiling reveals genetic linkage between peroxidase gene clusters and basal host and non-host resistance to rusts and mildew in barley. PLoS ONE 2010, 5, e10495. [Google Scholar] [CrossRef]
- Shtaya, M.J.Y.; Marcel, T.C.; Sillero, J.C.; Niks, R.E.; Rubiales, D. Identification of QTLs for powdery mildew and scald resistance in barley. Euphytica 2006, 151, 421–429. [Google Scholar] [CrossRef]
- Schweizer, P.; Stein, N. Large-scale data integration reveals colocalization of gene functional groups with meta-QTL for multiple disease resistance in barley. Mol. Plant Microbe Interact. 2011, 24, 1492–1501. [Google Scholar] [CrossRef] [PubMed]
- Hilbers, S.; Fischbeck, G.; Jahoor, A. Localization of the Laevigatum Resistance Gene MlLa against Powdery Mildew in the Barley Genome by the Use of RFLP Markers. Plant Breed. 1992, 109, 335–338. [Google Scholar] [CrossRef]
- Dros, J. The creation and maintenance of two spring barley varieties. Euphytica 1957, 6, 45–48. [Google Scholar] [CrossRef]
- Hoseinzadeh, P.; Ruge-Wehling, B.; Schweizer, P.; Stein, N.; Pidon, H. High Resolution Mapping of a Hordeum bulbosum-Derived Powdery Mildew Resistance Locus in Barley Using Distinct Homologous Introgression Lines. Front. Plant Sci. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Kurth, J.; Kolsch, R.; Simons, V.; Schulze-Lefert, P. A high-resolution genetic map and a diagnostic RFLP marker for the Mlg resistance locus to powdery mildew in barley. Theor. Appl. Genet. 2001, 102, 53–60. [Google Scholar] [CrossRef]
- Afzal, A.J.; Wood, A.J.; Lightfoot, D.A. Plant Receptor-Like Serine Threonine Kinases: Roles in Signaling and Plant Defense. Mol. Plant Microbe Interact. 2008, 21, 507–517. [Google Scholar] [CrossRef]
- Backes, G.; Schwarz, G.; Wenzel, G.; Jahoor, A. Comparison between QTL analysis of powdery mildew resistance in barley based on detached primary leaves and on field data. Plant Breed. 1996, 115, 419–421. [Google Scholar] [CrossRef]
- Silvar, C.; Perovic, D.; Scholz, U.; Casas, A.M.; Igartua, E.; Ordon, F. Fine mapping and comparative genomics integration of two quantitative trait loci controlling resistance to powdery mildew in a Spanish barley landrace. Theor. Appl. Genet. 2012, 124, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Silvar, C.; Perovic, D.; Nussbaumer, T.; Spannagl, M.; Usadel, B.; Casas, A.; Igartua, E.; Ordon, F. Towards Positional Isolation of Three Quantitative Trait Loci Conferring Resistance to Powdery Mildew in Two Spanish Barley Landraces. PLoS ONE 2013, 8, e67336. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Letter | Pedigree | Type | Country of Origin | Row Type |
|---|---|---|---|---|
| A | Ylitornion | Landrace | Finland | Six-rowed |
| B | GN 06075 | Breeding line | Norway | Six-rowed |
| C | Lavrans | Cultivar | Norway | Six-rowed |
| D | RGT Planet | Cultivar | France | Two-rowed |
| E | MBR 1012 | Landrace | Former Yugoslavia | Six-rowed |
| F | Iron | Cultivar | Poland | Two-rowed |
| G | JLB06-034 | Landrace | Jordan | Two-rowed |
| H | GN 09096 | Breeding line | Norway | Six-rowed |
| I | SJ 111998 | Breeding line | Denmark | Two-rowed |
| J | Chevron | Cultivar | Switzerland | Six-rowed |
| K | Olve | Cultivar | Norway | Two-rowed |
| L | Brage | Cultivar | Norway | Six-rowed |
| M | Krasnodarskij 35 | Cultivar | Soviet Union | Two-rowed |
| N | Nordic | Cultivar | US | Six-rowed |
| O | Fairytale | Cultivar | Denmark | Two-rowed |
| P | GN 09005 | Breeding line | Norway | Six-rowed |
| Q | Gaffelbyg | Landrace | Denmark | Six-rowed |
| Population | Crosses | No of Progenies |
|---|---|---|
| MAGIC 1 | ((A × B) × (C × D)) × ((E × F) × (G × H)) | 122 |
| MAGIC 2 | ((A × B) × (C × D)) × ((E × F) × (G × I)) | 29 |
| MAGIC 3 | ((J × D) × (K × L)) × ((M × F) × (N × O)) | 81 |
| MAGIC 4 | ((P × O) × (Q × O)) × ((M × F) × (N × O)) | 303 |
| Environment | Location | Entity | Year | Sowing Day | Replications | Observation Day |
|---|---|---|---|---|---|---|
| 1 | Dyngby | Nordic Seed | 2017 | 8 April | 2 | 1 June and 21 June |
| 2 | Dyngby | Nordic Seed | 2018 | 14 April | 2 | 25 June |
| 3 | Horsens | Sejet Plant Breeding | 2018 | 13 April | 1 | 25 May and 15 June |
| Chromosome | MAGIC 1 + 2 | MAGIC 3 | MAGIC 4 | MAGIC 1 to 4 |
|---|---|---|---|---|
| 1H | 15 | 60 | 56 | 17 |
| 2H | 14 | 40 | 23 | 17 |
| 3H | 7 | 44 | 28 | 15 |
| 4H | 12 | 45 | 34 | 16 |
| 5H | 13 | 31 | 29 | 19 |
| 6H | 12 | 140 | 429 | 20 |
| 7H | 19 | 33 | 497 | 32 |
| Genome-wide | 14 | 38 | 33 | 19 |
| QTL | SNP | Chromosome | Position (bp) | p-Value | LOD | MAF | Effect | MAGIC Population | Founder with pos. Allele | Lines with pos. Allele |
|---|---|---|---|---|---|---|---|---|---|---|
| Qrbg_1H_1 | JHI_Hv50k_2016_7757 | 1H | 6,976,974 | 6.06 × 109 | 8.218 | 0.306 | −0.376 | 1 + 2 | GN06075, GN09096, Iron, Lavrans | 37 |
| BOPA2_12_30918 | 1H | 8,935,905 | 1.45 × 1013 | 12.840 | 0.343 | - | 1 to 4 | Brage, Fairytale, GN06075, Iron, Krasnordarskij 35, Lavrans, SJ111998 | 315 | |
| JHI_Hv50k_2016_10019 | 1H | 9,240,411 | 1.63 × 1012 | 11.788 | 0.313 | −0.531 | 3 | Iron | 25 | |
| SCRI_RS_148733 | 1H | 10,236,703 | 1.55 × 1016 | 15.809 | 0.151 | 0.429 | 4 | Fairytale, GN09005, Iron, Krasnordarskij 35 | 233 | |
| Qrbg_1H_2 | JHI_Hv50k_2016_14683 | 1H | 18,358,190 | 1.19 × 108 | 7.926 | 0.233 | - | 1 to 4 | Chevron, Gaffelbyg, GN06075, GN09096, Iron, Lavrans, Nordic, Ylitornion | 101 |
| Qrbg_1H_3 | JHI_Hv50k_2016_37800 | 1H | 492,560,309 | 3.28 × 107 | 6.484 | 0.299 | −0.169 | 4 | Nordic | 82 |
| Qrbg_2H_1 | BOPA1_7623_818 | 2H | 52,014,030 | 2.29 × 105 | 4.640 | 0.147 | 0.204 | 4 | Krasnordarskij 35, Nordic | 39 |
| JHI_Hv50k_2016_87627 | 2H | 122,769,327 | 3.56 × 107 | 6.448 | 0.289 | −0.332 | 3 | Fairytale, Iron, Olve, RGT Planet | 20 | |
| Qrbg_2H_2 | JHI_Hv50k_2016_141795 | 2H | 754,879,170 | 3.34 × 1011 | 10.476 | 0.206 | −0.360 | 4 | Fairytale, GN09005, Iron | 57 |
| JHI_Hv50k_2016_142889 | 2H | 757,857,556 | 1.00 × 106 | 5.999 | 0.349 | 0.286 | 3 | Fairytale, Iron, Krasnordarskij 35, Olve, RGT Planet | 49 | |
| JHI_Hv50k_2016_147232 | 2H | 765,628,800 | 6.32 × 109 | 8.200 | 0.204 | - | 1 to 4 | Fairytale, JLB06-034 | 98 | |
| Qrbg_3H_1 | BOPA2_12_30192 | 3H | 21,019,740 | 1.46 × 106 | 5.835 | 0.276 | −0.258 | 1 + 2 | GN06075, GN09096, JLB06-034, Lavrans, RGT Planet, SJ111998 | 91 |
| Qrbg_4H_1 | JHI_Hv50k_2016_230367 | 4H | 12,239,110 | 9.93 × 106 | 5.003 | 0.253 | −0.285 | 3 | Brage, Chevron, Fairytale, Krasnordarskij 35, RGT Planet | 16 |
| Qrbg_4H_2 | JHI_Hv50k_2016_265870 | 4H | 621,555,275 | 6.47 × 1033 | 32.189 | 0.358 | −1.338 | 1 + 2 | RGT Planet, SJ111998 | 46 |
| 3.24 × 1027 | 26.489 | 0.102 | - | 1 to 4 | GN09005, RGT Planet, SJ111998 | 47 | ||||
| JHI_Hv50k_2016_267153 | 4H | 623,655,675 | 6.28 × 107 | 6.202 | 0.397 | - | 1 to 4 | Gaffelbyg, GN09005, Iron, JLB06-034, RGT Planet, SJ111998, Ylitornion | 187 | |
| Qrbg_5H_1 | JHI_Hv50k_2016_276999 | 5H | 396,682 | 3.26 × 106 | 5.487 | 0.403 | −0.316 | 1 + 2 | JLB06-034, RGT Planet, SJ111998 | 51 |
| Qrbg_5H_2 | SCRI_RS_166374 | 5H | 272,385,765 | 7.58 × 107 | 6.120 | 0.289 | 0.388 | 3 | Brage, Fairytale, Iron, RGT Planet | 57 |
| Qrbg_7H_1 | JHI_Hv50k_2016_444783 | 7H | 9,411,745 | 2.97 × 1022 | 21.528 | 0.065 | - | 1 to 4 | Gaffelbyg | 31 |
| JHI_Hv50k_2016_449746 | 7H | 14,735,678 | 1.40 × 1026 | 25.855 | 0.118 | −0.832 | 4 | Gaffelbyg | 32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novakazi, F.; Krusell, L.; Jensen, J.D.; Orabi, J.; Jahoor, A.; Bengtsson, T.; on behalf of the PPP Barley Consortium. You Had Me at “MAGIC”!: Four Barley MAGIC Populations Reveal Novel Resistance QTL for Powdery Mildew. Genes 2020, 11, 1512. https://doi.org/10.3390/genes11121512
Novakazi F, Krusell L, Jensen JD, Orabi J, Jahoor A, Bengtsson T, on behalf of the PPP Barley Consortium. You Had Me at “MAGIC”!: Four Barley MAGIC Populations Reveal Novel Resistance QTL for Powdery Mildew. Genes. 2020; 11(12):1512. https://doi.org/10.3390/genes11121512
Chicago/Turabian StyleNovakazi, Fluturë, Lene Krusell, Jens Due Jensen, Jihad Orabi, Ahmed Jahoor, Therése Bengtsson, and on behalf of the PPP Barley Consortium. 2020. "You Had Me at “MAGIC”!: Four Barley MAGIC Populations Reveal Novel Resistance QTL for Powdery Mildew" Genes 11, no. 12: 1512. https://doi.org/10.3390/genes11121512
APA StyleNovakazi, F., Krusell, L., Jensen, J. D., Orabi, J., Jahoor, A., Bengtsson, T., & on behalf of the PPP Barley Consortium. (2020). You Had Me at “MAGIC”!: Four Barley MAGIC Populations Reveal Novel Resistance QTL for Powdery Mildew. Genes, 11(12), 1512. https://doi.org/10.3390/genes11121512