The Reversion of cg05575921 Methylation in Smoking Cessation: A Potential Tool for Incentivizing Healthy Aging


 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Approval
2.2. Study Participants
2.3. Laboratory Measures
2.4. Statistical Analyses
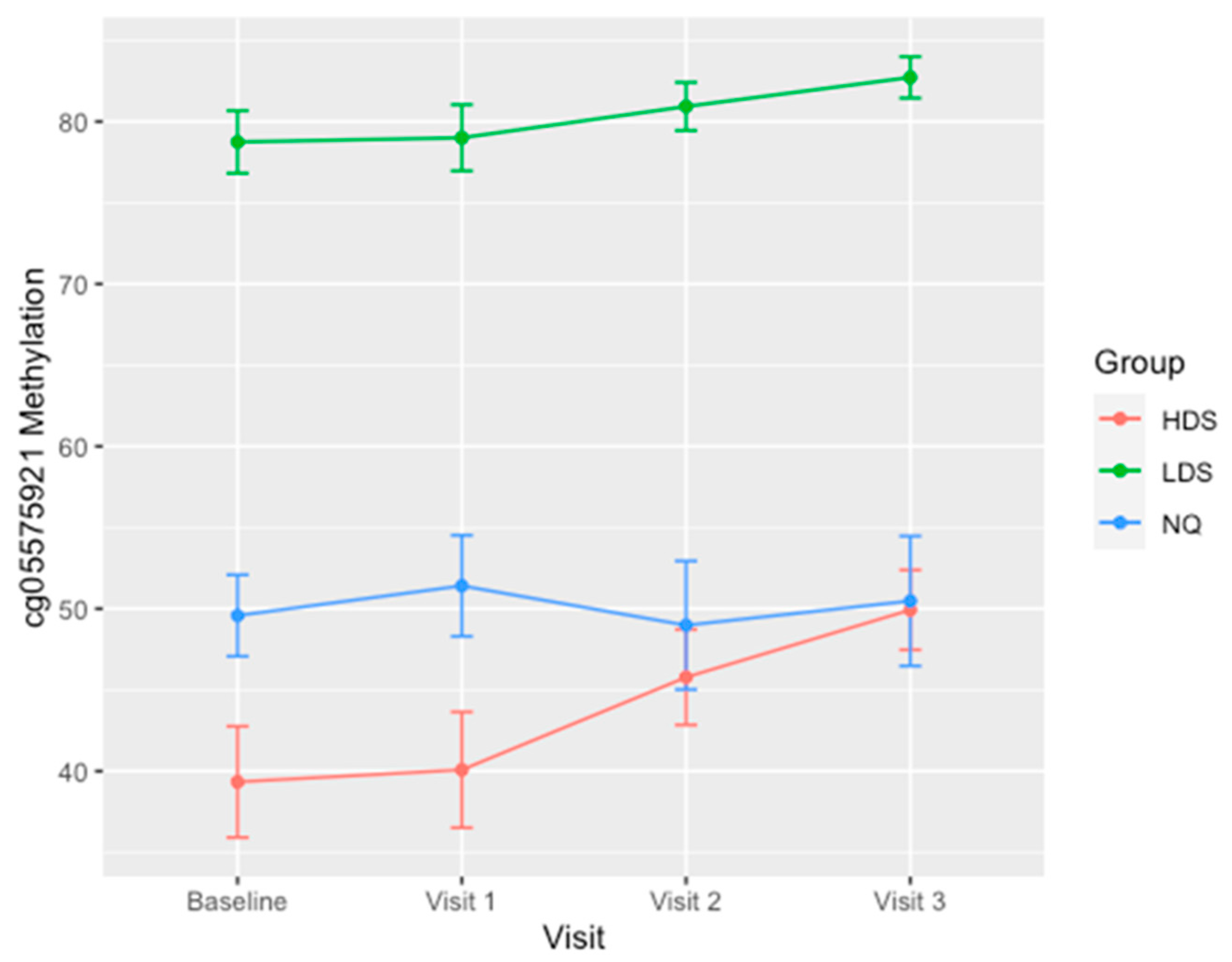
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Soriano-Tárraga, C.; Jiménez-Conde, J.; Giralt-Steinhauer, E.; Mola-Caminal, M.; Vivanco-Hidalgo, R.M.; Ois, A.; Rodríguez-Campello, A.; Cuadrado-Godia, E.; Sayols-Baixeras, S.; Elosua, R.; et al. Epigenome-wide association study identifies txnip gene associated with type 2 diabetes mellitus and sustained hyperglycemia. Hum. Mol. Genet. 2015, 25, 609–619. [Google Scholar] [CrossRef]
- Dayeh, T.; Tuomi, T.; Almgren, P.; Perfilyev, A.; Jansson, P.-A.; de Mello, V.D.; Pihlajamäki, J.; Vaag, A.; Groop, L.; Nilsson, E. DNA methylation of loci within abcg1 and phospho1 in blood DNA is associated with future type 2 diabetes risk. Epigenetics 2016, 11, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.C.; Loh, M.; Lehne, B.; Drong, A.; Kriebel, J.; Motta, V.; Wahl, S.; Elliott, H.R.; Rota, F.; Scott, W.R. Epigenome-wide association of DNA methylation markers in peripheral blood from indian asians and europeans with incident type 2 diabetes: A nested case-control study. Lancet Diabetes Endocrinol. 2015, 3, 526–534. [Google Scholar] [CrossRef]
- Vasanthakumar, A.; Davis, J.W.; Idler, K.; Waring, J.F.; Asque, E.; Riley-Gillis, B.; Grosskurth, S.; Srivastava, G.; Kim, S.; Nho, K. Harnessing peripheral DNA methylation differences in the alzheimer’s disease neuroimaging initiative (adni) to reveal novel biomarkers of disease. Clin. Epigenet. 2020, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Monick, M.M.; Beach, S.R.; Plume, J.; Sears, R.; Gerrard, M.; Brody, G.H.; Philibert, R.A. Coordinated changes in ahrr methylation in lymphoblasts and pulmonary macrophages from smokers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159B, 141–151. [Google Scholar] [CrossRef]
- Philibert, R.A.; Penaluna, B.; White, T.; Shires, S.; Gunter, T.; Liesveld, J.; Erwin, C.; Hollenbeck, N.; Osborn, T. A pilot examination of the genome-wide DNA methylation signatures of subjects entering and exiting short-term alcohol dependence treatment programs. Epigenetics 2014, 9, 1212–1219. [Google Scholar] [CrossRef]
- Wang, X.; Pan, Y.; Zhu, H.; Hao, G.; Huang, Y.; Barnes, V.; Shi, H.; Snieder, H.; Pankow, J.; North, K. An epigenome-wide study of obesity in african american youth and young adults: Novel findings, replication in neutrophils, and relationship with gene expression. Clin. Epigenet. 2018, 10, 1–9. [Google Scholar] [CrossRef]
- McDade, T.W.; Ryan, C.P.; Jones, M.J.; Hoke, M.K.; Borja, J.; Miller, G.E.; Kuzawa, C.W.; Kobor, M.S. Genome-wide analysis of DNA methylation in relation to socioeconomic status during development and early adulthood. Am. J. Phys. Anthropol. 2019, 169, 3–11. [Google Scholar] [CrossRef]
- Philibert, R.; Long, J.; Mill, J.; Beach, S.R.; Gibbons, F.X.; Gerrard, M.; Simons, R.; Pinho, P.; Ingle, D.; Dawes, K.; et al. A simple, rapid, interpretable, actionable and implementable digital pcr based mortality index. Epigenetics 2020, 1–15. [Google Scholar] [CrossRef]
- US Department of Health Human Services. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General; US Department of Health and Human Services: Washington, DC, USA; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2014.
- Gao, X.; Zhang, Y.; Breitling, L.P.; Brenner, H. Relationship of tobacco smoking and smoking-related DNA methylation with epigenetic age acceleration. Oncotarget 2016, 7, 46878. [Google Scholar] [CrossRef]
- Beach, S.R.H.; Dogan, M.V.; Lei, M.-K.; Cutrona, C.E.; Gerrard, M.; Gibbons, F.X.; Simons, R.L.; Brody, G.H.; Philibert, R.A. Methylomic aging as a window onto the influence of lifestyle: Tobacco and alcohol use alter the rate of biological aging. J. Am. Geriatr. Soc. 2015, 63, 2519–2525. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gao, X.; Just, A.C.; Colicino, E.; Wang, C.; Coull, B.A.; Hou, L.; Zheng, Y.; Vokonas, P.; Schwartz, J. Smoking-related DNA methylation is associated with DNA methylation phenotypic age acceleration: The veterans affairs normative aging study. Int. J. Environ. Res. Public Health 2019, 16, 2356. [Google Scholar] [CrossRef] [PubMed]
- Philibert, R.; Dogan, M.; Beach, S.R.H.; Mills, J.A.; Long, J.D. Ahrr methylation predicts smoking status and smoking intensity in both saliva and blood DNA. Am. J. Genet. 2019, 183, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Guida, F.; Campanella, G.; Castagné, R.; Chadeau-Hyam, M.; Vermeulen, R.C.H.; Vineis, P.; Georgiadis, P.; Kyrtopoulos, S.A.; Lund, E.; Sandanger, T.M.; et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum. Mol. Genet. 2015, 24, 2349–2359. [Google Scholar] [CrossRef]
- Wilson, R.; Wahl, S.; Pfeiffer, L.; Ward-Caviness, C.K.; Kunze, S.; Kretschmer, A.; Reischl, E.; Peters, A.; Gieger, C.; Waldenberger, M. The dynamics of smoking-related disturbed methylation: A two time-point study of methylation change in smokers, non-smokers and former smokers. BMC Genom. 2017, 18, 805. [Google Scholar] [CrossRef]
- McCartney, D.L.; Stevenson, A.J.; Hillary, R.F.; Walker, R.M.; Bermingham, M.L.; Morris, S.W.; Clarke, T.-K.; Campbell, A.; Murray, A.D.; Whalley, H.C.; et al. Epigenetic signatures of starting and stopping smoking. EBioMedicine 2018, 37, 214–220. [Google Scholar] [CrossRef]
- Philibert, R.; Hollenbeck, N.; Andersen, E.; McElroy, S.; Wilson, S.; Vercande, K.; Beach, S.R.; Osborn, T.; Gerrard, M.; Gibbons, F.X.; et al. Reversion of ahrr demethylation is a quantitative biomarker of smoking cessation. Front. Psychiatry 2016, 7, 55. [Google Scholar] [CrossRef]
- Mills, J.A.; Beach, S.R.; Dogan, M.; Simons, R.L.; Gibbons, F.X.; Long, J.D.; Philibert, R. A direct comparison of the relationship of epigenetic aging and epigenetic substance consumption markers to mortality in the framingham heart study. Genes 2019, 10, 51. [Google Scholar] [CrossRef]
- Parrott, S.; Godfrey, C. Econ of smoking cess. BMJ 2004, 328, 947–949. [Google Scholar] [CrossRef]
- Öberg, M.; Jaakkola, M.S.; Woodward, A.; Peruga, A.; Prüss-Ustün, A. Worldwide burden of disease from exposure to second-hand smoke: A retrospective analysis of data from 192 countries. Lancet 2011, 377, 139–146. [Google Scholar] [CrossRef]
- Stitzer, M.L.; Vandrey, R. Contingency management: Utility in the treatment of drug abuse disorders. Clin. Pharm. Ther. 2008, 83, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Cooney, J.L.; Cooper, S.; Grant, C.; Sevarino, K.; Krishnan-Sarin, S.; Gutierrez, I.A.; Cooney, N.L. A randomized trial of contingency management for smoking cessation during intensive outpatient alcohol treatment. J. Subst. Abuse Treat. 2017, 72, 89–96. [Google Scholar] [CrossRef]
- Dallery, J.; Raiff, B.R.; Kim, S.J.; Marsch, L.A.; Stitzer, M.; Grabinski, M.J. Nationwide access to an internet-based contingency management intervention to promote smoking cessation: A randomized controlled trial. Addiction 2017, 112, 875–883. [Google Scholar] [CrossRef]
- Etter, J.-F.; Schmid, F. Effects of large financial incentives for long-term smoking cessation: A randomized trial. J. Am. Coll. Cardiol. 2016, 68, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Secades-Villa, R.; García-Rodríguez, O.; López-Núñez, C.; Alonso-Pérez, F.; Fernández-Hermida, J.R. Contingency management for smoking cessation among treatment-seeking patients in a community setting. Drug Alcohol Depend. 2014, 140, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Notley, C.; Gentry, S.; Livingstone-Banks, J.; Bauld, L.; Perera, R.; Hartmann-Boyce, J. Incentives for smoking cessation. Cochrane Datab. Syst. Rev. 2019. [Google Scholar] [CrossRef]
- Prendergast, M.; Podus, D.; Finney, J.; Greenwell, L.; Roll, J. Contingency management for treatment of substance use disorders: A meta-analysis. Addiction 2006, 101, 1546–1560. [Google Scholar] [CrossRef]
- Halpern, S.D.; French, B.; Small, D.S.; Saulsgiver, K.; Harhay, M.O.; Audrain-McGovern, J.; Loewenstein, G.; Brennan, T.A.; Asch, D.A.; Volpp, K.G. Randomized trial of four financial-incentive programs for smoking cessation. NEJM 2015, 372, 2108–2117. [Google Scholar] [CrossRef]
- Jacobs, D.R., Jr.; Adachi, H.; Mulder, I.; Kromhout, D.; Menotti, A.; Nissinen, A.; Blackburn, H. Cigarette smoking and mortality risk: Twenty-five–year follow-up of the seven countries study. Arch. Intern. Med. 1999, 159, 733–740. [Google Scholar] [CrossRef]
- Florescu, A.; Ferrence, R.; Einarson, T.; Selby, P.; Soldin, O.; Koren, G. Methods for quantification of exposure to cigarette smoking and environmental tobacco smoke: Focus on developmental toxicology. Ther. Drug Monit. 2009, 31, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Philibert, R.; Dogan, M.; Noel, A.; Miller, S.; Krukow, B.; Papworth, E.; Cowley, J.; Long, J.D.; Beach, S.R.; Black, D.W. Dose response and prediction characteristics of a methylation sensitive digital pcr assay for cigarette consumption in adults. Front. Genet. 2018, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Dawes, K.; Andersen, A.; Papworth, E.; Hundley, B.; Hutchens, N.; El Manawy, H.; Becker, A.; Sampson, L.; Philibert, W.; Gibbons, F.X. Refinement of cg05575921 demethylation response in nascent smoking. Clin. Epigenet. 2020, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Heatherton, T.F.; Kozlowski, L.T.; Frecker, R.C.; Fagerstrom, K.O. The fagerstrom test for nicotine dependence: A revision of the fagerstrom tolerance questionnaire. Br. J. Addict. 1991, 86, 1119–1127. Available online: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=1932883 (accessed on 5 May 2020). [CrossRef] [PubMed]
- Lahiri, D.K.; Nurnberger, J.I., Jr. A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res. 1991, 19, 5444. [Google Scholar] [CrossRef] [PubMed]
- Kramer, C.Y. Extension of multiple range tests to group means with unequal numbers of replications. Biometrics 1956, 12, 307–310. [Google Scholar] [CrossRef]
- Finucane, M.M.; Samet, J.H.; Horton, N.J. Translational methods in biostatistics: Linear mixed effect regression models of alcohol consumption and hiv disease progression over time. Epidemiol. Perspect. Innov. 2007, 4, 8. [Google Scholar] [CrossRef]
- Andersen, A.M.; Philibert, R.A.; Gibbons, F.X.; Simons, R.L.; Long, J. Accuracy and utility of an epigenetic biomarker for smoking in populations with varying rates of false self-report. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017, 174, 641–650. [Google Scholar] [CrossRef]
- Andersen, A.; Reimer, R.; Dawes, K.; Becker, A.; Hutchens, N.; Miller, S.; Dogan, M.V.; Hundley, B.; Mills, J.A.; Long, J.; et al. DNA methylation differentiates smoking from vaping and non-combustible tobacco use. 2020; in submission. [Google Scholar]
- Gan, W.Q.; Cohen, S.B.; Man, S.F.; Sin, D.D. Sex-related differences in serum cotinine concentrations in daily cigarette smokers. Nicotine Tob. Res. 2008, 10, 1293–1300. [Google Scholar] [CrossRef]
- Gibbons, F.X.; Eggleston, T.J. Smoker networks and the typical smoker: A prospective analysis of smoking cessation. Health Psychol. 1996, 15, 469. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| ALL SUBJECTS | NON-QUITTERS | QUITTERS | ||
|---|---|---|---|---|
| HDS | LDS | |||
| N TOTAL COUNT | 67 | 47 | 11 | 9 |
| SEX COUNT | ||||
| FEMALE | 32 | 23 | 3 | 6 |
| MALE | 35 | 24 | 8 | 3 |
| AGE (YEARS) | 43.7 ± 10.0 | 45.4 ± 9.6 | 40.3 ± 11.1 | 39.1 ± 8.9 |
| ETHNICITY COUNT | ||||
| WHITE | 62 | 43 | 11 | 8 |
| HISPANIC WHITE | 1 | - | - | 1 |
| AFRICAN AMERICAN | 2 | 2 | - | - |
| ASIAN | 2 | 2 | - | - |
| PACK YEAR LIFETIME | 28.0 ± 18.1 | 30.6 ± 20.3 | 25.1 ± 10.3 | 18.5 ± 7.8 |
| CIGARETTES/DAY LAST MONTH | 17.6 ± 8.7 | 18.4 ± 9.6 | 18.4 ± 5.8 | 12.1 ± 2.3 |
| FTND SCORE | 3.8 ± 2.2 | 3.9 ± 2.4 | 4.2 ±2.2 | 3.2 ±1.3 |
| INTAKE COT (NG/ML) | 251 ± 110 | 270 ± 112 † | 255 ± 78 | 146 ± 74 |
| INTAKE CG05575921 | 51.6 ± 18.8 | 49.3 ± 16.9 †† | 39.3 ±11.4 †† | 78.7 ± 5.8 |
| Baseline to Visit 1 | Visit 1 to Visit 2 | Visit 2 to Visit 3 | ||||
|---|---|---|---|---|---|---|
| HDS | LDS | HDS | LDS | HDS | LDS | |
| Reversion | ||||||
| Total number | 7 | 6 | 8 | 7 | 10 | 9 |
| Mean Δβ | 2.13 ± 1.47% | 2.55 ± 2.19% | 5.38 ± 2.48% | 2.63 ± 1.77% | 4.36 ± 2.71% | 1.79 ± 1.70% |
| Demethylated | ||||||
| Total number | 3 | 3 | 1 | 2 | - | - |
| Mean Δβ | −3.30 ± 3.38% | −3.47 ± 1.94% | −1.40% | −0.54% | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Philibert, R.; Mills, J.A.; Long, J.D.; Salisbury, S.E.; Comellas, A.; Gerke, A.; Dawes, K.; Vander Weg, M.; Hoffman, E.A. The Reversion of cg05575921 Methylation in Smoking Cessation: A Potential Tool for Incentivizing Healthy Aging. Genes 2020, 11, 1415. https://doi.org/10.3390/genes11121415
Philibert R, Mills JA, Long JD, Salisbury SE, Comellas A, Gerke A, Dawes K, Vander Weg M, Hoffman EA. The Reversion of cg05575921 Methylation in Smoking Cessation: A Potential Tool for Incentivizing Healthy Aging. Genes. 2020; 11(12):1415. https://doi.org/10.3390/genes11121415
Chicago/Turabian StylePhilibert, Robert, James A. Mills, Jeffrey D. Long, Sue Ellen Salisbury, Alejandro Comellas, Alicia Gerke, Kelsey Dawes, Mark Vander Weg, and Eric A. Hoffman. 2020. "The Reversion of cg05575921 Methylation in Smoking Cessation: A Potential Tool for Incentivizing Healthy Aging" Genes 11, no. 12: 1415. https://doi.org/10.3390/genes11121415
APA StylePhilibert, R., Mills, J. A., Long, J. D., Salisbury, S. E., Comellas, A., Gerke, A., Dawes, K., Vander Weg, M., & Hoffman, E. A. (2020). The Reversion of cg05575921 Methylation in Smoking Cessation: A Potential Tool for Incentivizing Healthy Aging. Genes, 11(12), 1415. https://doi.org/10.3390/genes11121415