Genetic Potential of the Biocontrol Agent Pseudomonas brassicacearum (Formerly P. trivialis) 3Re2-7 Unraveled by Genome Sequencing and Mining, Comparative Genomics and Transcriptomics

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Results and Discussion
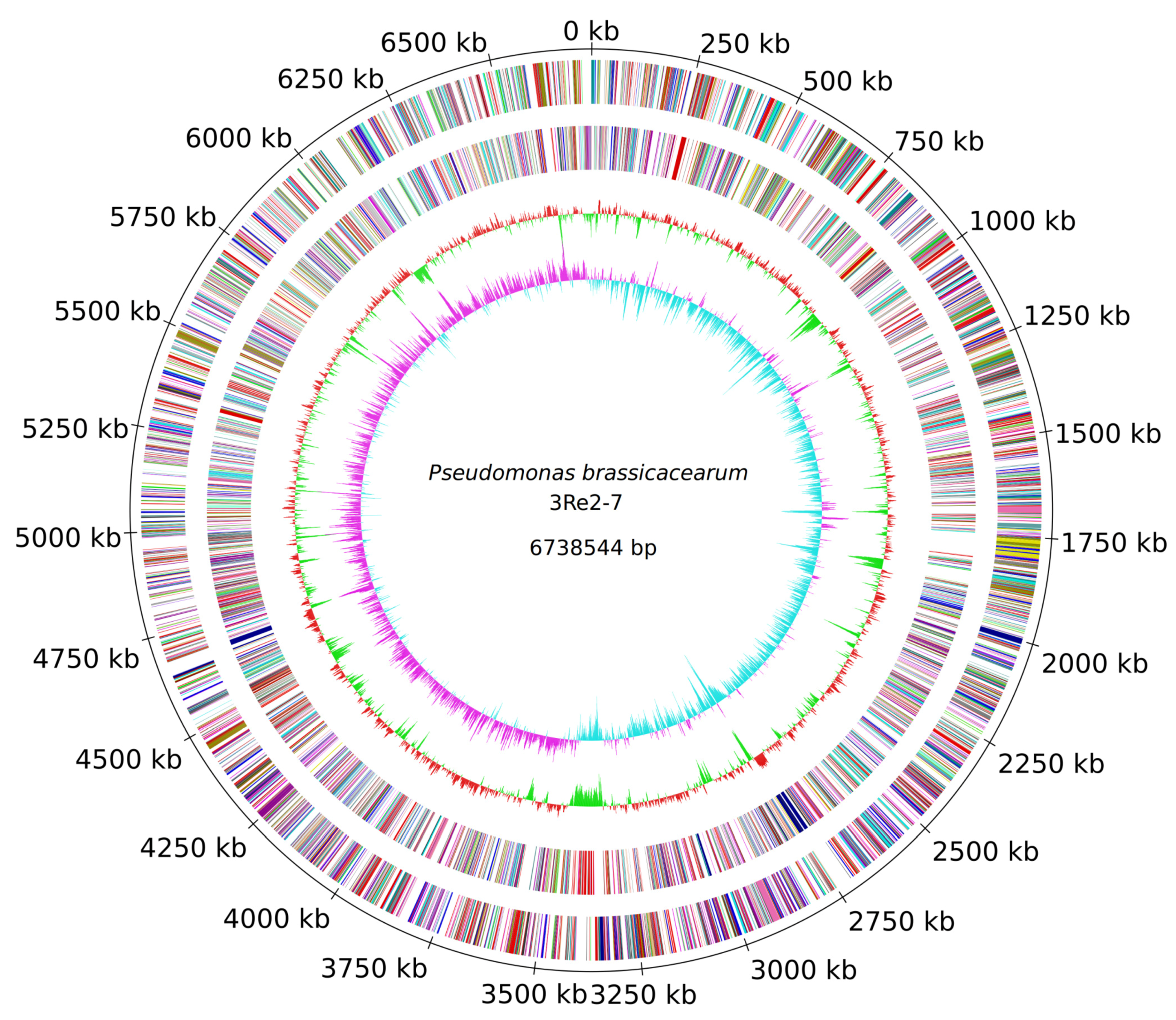
2.1. Genome Sequencing and General Genome Features of P. brassicacearum 3Re2-7
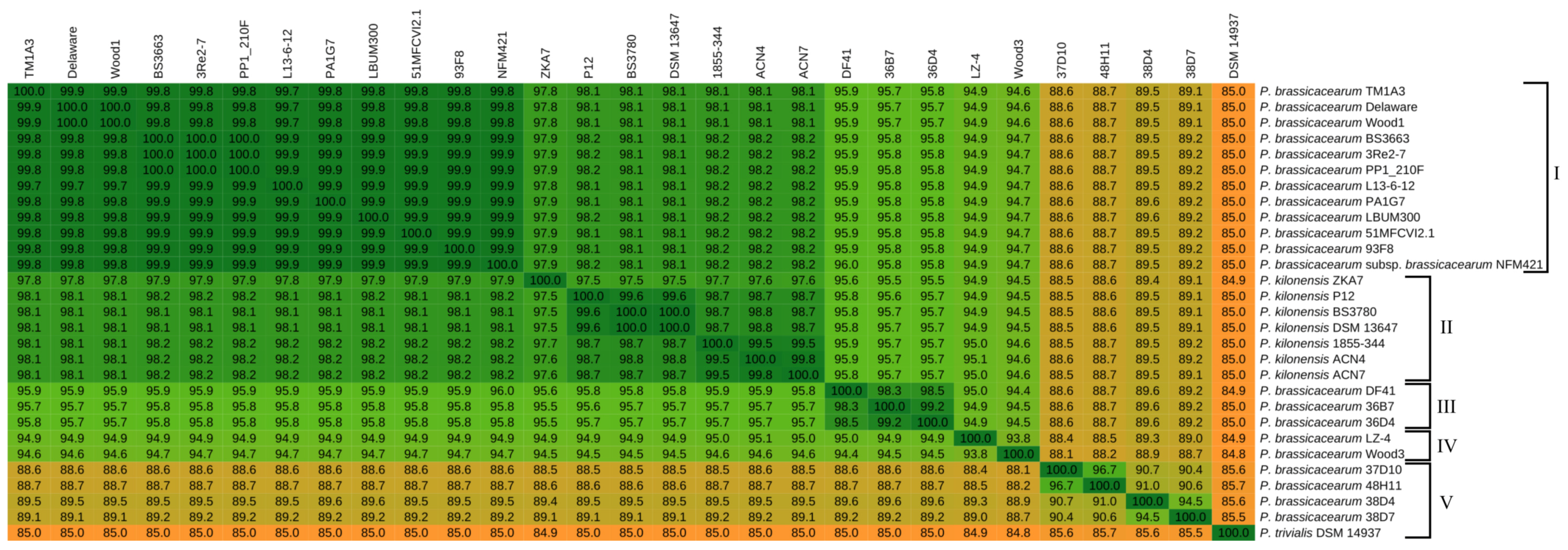
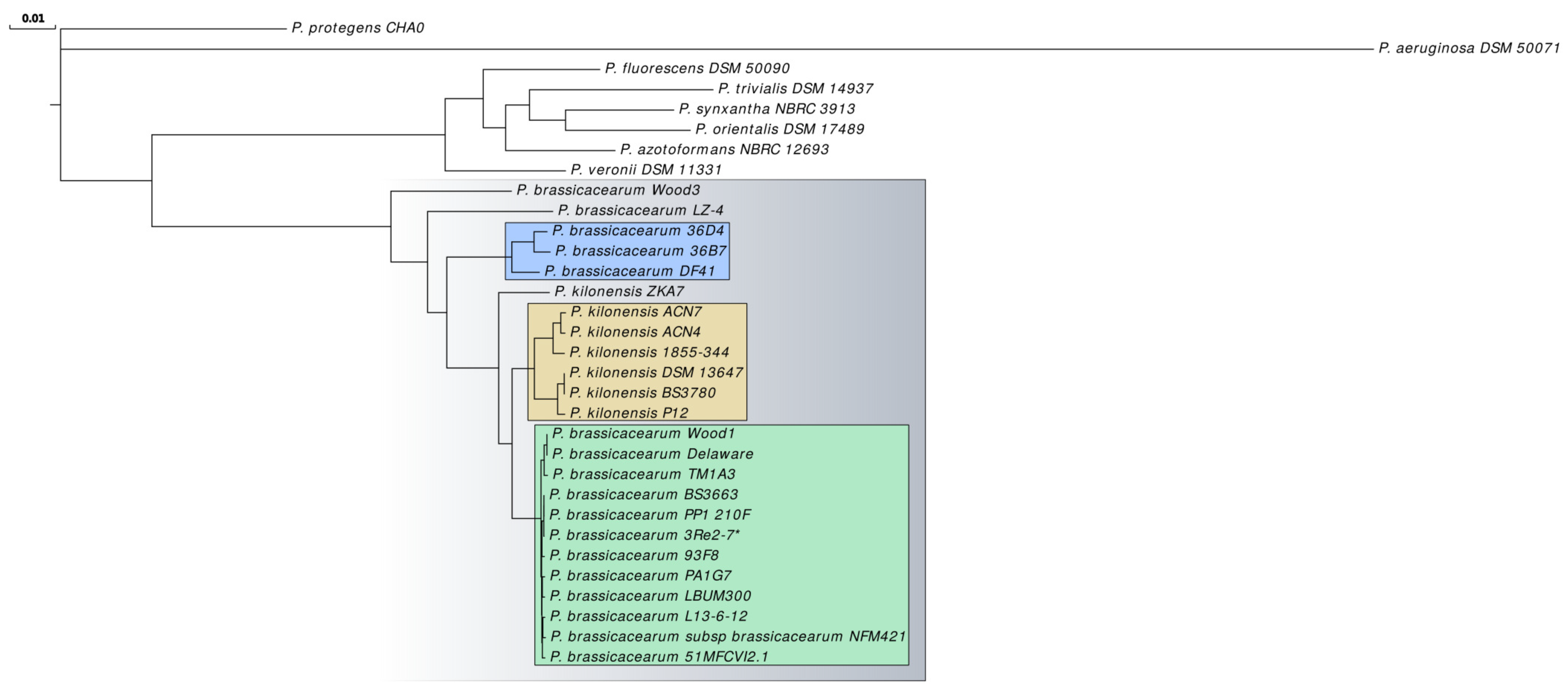
2.2. Taxonomic Classification Based on Whole-Genome and Core Gene Analyses
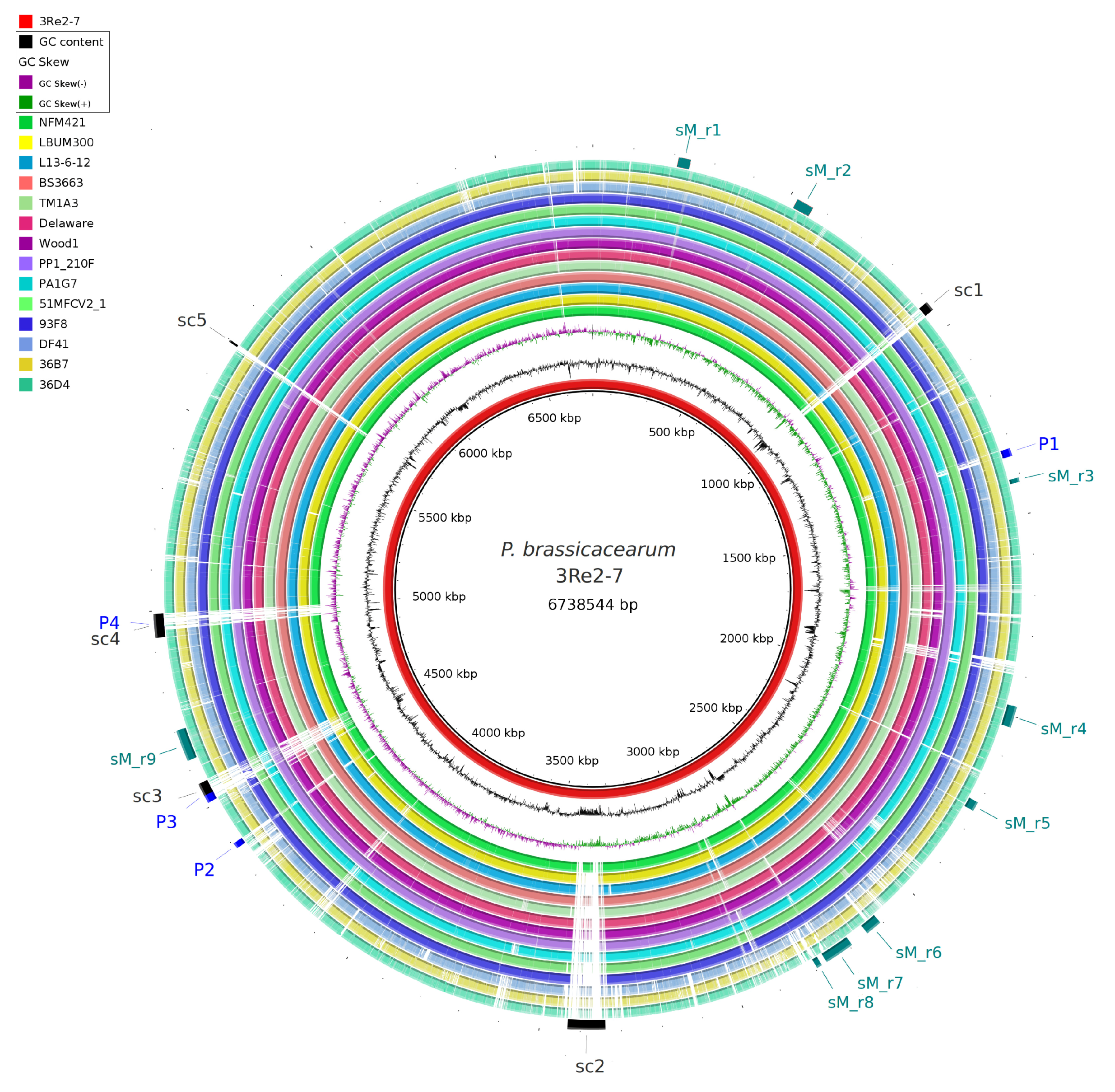
2.3. Comparative Genomics Revealed Genes Unique to P. brassicacearum 3Re2-7
2.4. Genome Mining of P. brassicacearum 3Re2-7 Revealed Biocontrol Determinants
2.4.1. Secondary Metabolism and Antibiotics
2.4.2. Induced Systemic Resistance and Plant Growth Promotion
2.4.3. Pathogen Inhibition
2.4.4. Other Biocontrol Features
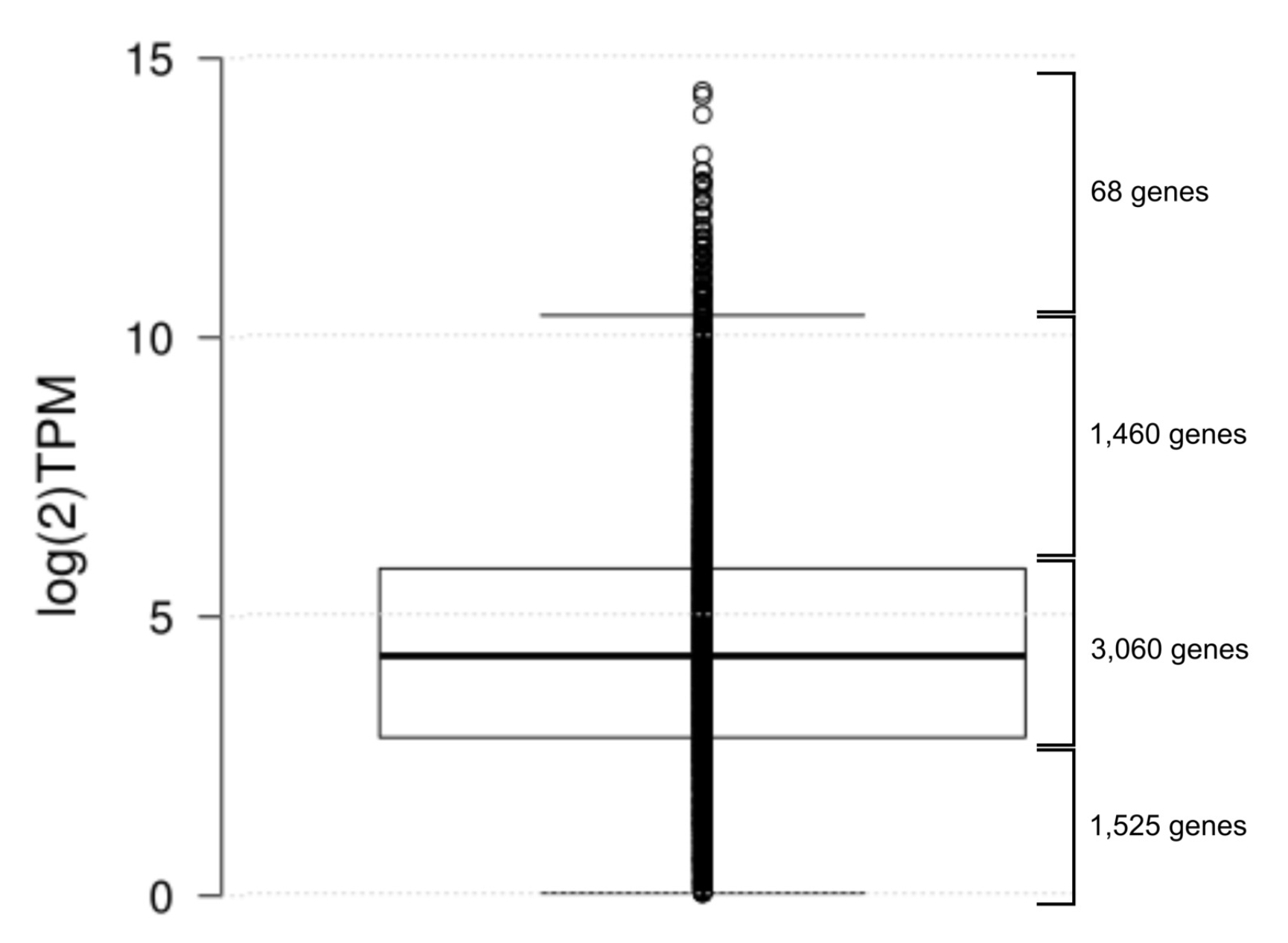
2.5. Transcriptome Sequencing of P. brassicacearum 3Re2-7 Confirms Expression of Biocontrol Related Genes
3. Materials and Methods
3.1. PacBio Library Preparation, Sequencing and Genome Assembly
3.2. Genome Annotation and Mining
3.3. Comparative Analysis
3.4. Transcriptome Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hartmann, M.; Frey, B.; Mayer, J.; Mäder, P.; Widmer, F. Distinct soil microbial diversity under long-term organic and conventional farming. ISME J. 2015, 9, 1177–1194. [Google Scholar] [CrossRef] [PubMed]
- Oerke, E.C. Crop Losses to Pests. J. Agric. Sci. 2006, 144, 31–43. [Google Scholar] [CrossRef]
- Berg, G.; Köberl, M.; Rybakova, D.; Müller, H.; Grosch, R.; Smalla, K. Plant Microbial Diversity Is Suggested as the Key to Future Biocontrol and Health Trends. FEMS Microbiol Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.P.; Dietel, K.; Rändler, M.; Schmid, M.; Junge, H.; Borriss, R.; Hartmann, A.; Grosch, R. Effects of Bacillus amyloliquefaciens FZB42 on Lettuce Growth and Health under Pathogen Pressure and Its Impact on the Rhizosphere Bacterial Community. PLoS ONE 2013, 8, e68818. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Sanz, D.; Meier-Kolthoff, J.P.; Göker, M.; Martín, M.; Rivilla, R.; Redondo-Nieto, M. Genomic and genetic diversity within the Pseudomonas fluoresces complex. PLoS ONE 2016, 11, e0150183. [Google Scholar]
- Pieterse, C.M.; Zamioudis, C.; Berendsen, R.L.; Weller, D.M.; Van Wees, S.C.; Bakker, P.A. Induced Systemic Resistance by Beneficial Microbes. Annu. Rev. Phytopathol. 2014, 52, 347–375. [Google Scholar] [CrossRef] [PubMed]
- Maksimov, I.V.; Abizgil’dina, R.R.; Pusenkova, L.I. Plant growth promoting rhizobacteria as alternative to chemical crop protectors from pathogens (review). Appl. Biochem. Microbiol. 2011, 47, 333–345. [Google Scholar] [CrossRef]
- Paterson, J.; Jahanshah, G.; Li, Y.; Wang, Q.; Mehnaz, S.; Gross, H. The contribution of genome mining strategies to the understanding of active principles of PGPR strains. FEMS Microbiol. Ecol. 2017, 93, fiw249. [Google Scholar] [CrossRef]
- Nelkner, J.; Henke, C.; Lin, T.W.; Pätzold, W.; Hassa, J.; Jaenicke, S.; Grosch, R.; Pühler, A.; Sczyrba, A.; Schlüter, A. Effect of Long-Term Farming Practices on Agricultural Soil Microbiome Members Represented by Metagenomically Assembled Genomes (MAGs) and Their Predicted Plant-Beneficial Genes. Genes 2019, 10, 424. [Google Scholar] [CrossRef]
- Mishra, J.; Arora, N.K. Secondary metabolites of fluorescent pseudomonads in biocontrol of phytopathogens for sustainable agriculture. Appl. Soil Ecol. 2018, 125, 35–45. [Google Scholar] [CrossRef]
- Couillerot, O.; Prigent-Combaret, C.; Caballero-Mellado, J.; Moënne-Loccoz, Y. Pseudomonas fluorescens and closely-related fluorescent pseudomonads as biocontrol agents of soil-borne phytopathogens. Lett. Appl. Microbiol. 2009, 48, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Barret, M.; Frey-Klett, P.; Boutin, M.; Guillerm-Erckelboudt, A.Y.; Martin, F.; Guillot, L.; Sarniguet, A. The plant pathogenic fungus Gaeumannomyces graminis var. tritici improves bacterial growth and triggers early gene regulations in the biocontrol strain Pseudomonas fluorescens Pf29Arp. New Phytol. 2009, 181, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Hernández-León, R.; Rojas-Solís, D.; Contreras-Pérez, M.; Orozco-Mosqueda, M.d.C.; Macías-Rodríguez, L.I.; Reyes-de la Cruz, H.; Valencia-Cantero, E.; Santoyo, G. Characterization of the antifungal and plant growth-promoting effects of diffusible and volatile organic compounds produced by Pseudomonas fluorescens strains. Biol. Control 2015, 81, 83–92. [Google Scholar] [CrossRef]
- Michelsen, C.F.; Watrous, J.; Glaring, M.A.; Kersten, R.; Koyama, N.; Dorrestein, P.C.; Stougaard, P. Nonribosomal peptides, key biocontrol components for Pseudomonas fluorescens in5, isolated from a Greenlandic suppressive soil. mBio 2015, 6, e00079. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, R.C.; Glaring, M.A.; Olsson, S.; Stougaard, P. Transcriptomic profiling of microbe–microbe interactions reveals the specific response of the biocontrol strain P. fluorescens In5 to the phytopathogen Rhizoctonia solani. BMC Res. Notes 2017, 10, 376. [Google Scholar] [CrossRef] [PubMed]
- Achouak, W.; Sutra, L.; Heulin, T.; Meyer, J.M.; Fromin, N.; Degraeve, S.; Christen, R.; Gardan, L. Pseudomonas brassicacearum sp. nov. and Pseudomonas thivervalensis sp. nov., two root-associated bacteria isolated from Brassica napus and Arabidopsis thaliana. Int. J. Syst. Evol. Microbiol. 2000, 50, 9–18. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, D.; Li, C.; Sun, Q.; Li, L.; Liu, F.; Shen, Q.; Shen, B. Isolation and characterization of Pseudomonas brassicacearum J12 as an antagonist against Ralstonia solanacearum and identification of its antimicrobial components. Microbiol. Res. 2012, 167, 388–394. [Google Scholar] [CrossRef]
- Ortet, P.; Barakat, M.; Lalaouna, D.; Fochesato, S.; Barbe, V.; Vacherie, B.; Santaella, C.; Heulin, T.; Achouak, W. Complete genome sequence of a beneficial plant root-associated bacterium, Pseudomonas brassicacearum. J. Bacteriol. 2011, 193, 3146. [Google Scholar] [CrossRef]
- Loewen, P.C.; Switala, J.; Fernando, W.G.D.; de Kievit, T. Genome Sequence of Pseudomonas brassicacearum DF41. Genome Announc. 2014, 2, 10–11. [Google Scholar] [CrossRef]
- Khayi, S.; des Essarts, Y.R.; Mondy, S.; Moumni, M.; Hélias, V.; Beury-Cirou, A.; Faure, D. Draft Genome Sequences of the Three Pectobacterium-Antagonistic Bacteria Pseudomonas brassicacearum PP1-210F and PA1G7 and Bacillus simplex BA2H3. Genome Announc. 2015, 3, e01497-14. [Google Scholar] [CrossRef]
- Zachow, C.; Müller, H.; Monk, J.; Berg, G. Complete genome sequence of Pseudomonas brassicacearum strain L13-6-12, a biological control agent from the rhizosphere of potato. Stand. Genom. Sci. 2017, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Novinscak, A.; Gadkar, V.J.; Joly, D.L.; Filion, M. Complete Genome Sequence of Pseudomonas brassicacearum LBUM300, a Disease-Suppressive Bacterium with Antagonistic Activity toward Fungal, Oomycete, and Bacterial Plant Pathogens. Genome Announc. 2016, 4, 2012–2013. [Google Scholar] [CrossRef] [PubMed]
- Lanteigne, C.; Gadkar, V.J.; Wallon, T.; Novinscak, A.; Filion, M. Production of DAPG and HCN by Pseudomonas sp. LBUM300 contributes to the biological control of bacterial canker of tomato. Phytopathology 2012, 102, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Kai, M.; Effmert, U.; Berg, G.; Piechulla, B. Volatiles of bacterial antagonists inhibit mycelial growth of the plant pathogen Rhizoctonia solani. Arch. Microbiol. 2007, 187, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Krechel, A.; Ditz, M.; Sikora, R.A.; Ulrich, A.; Hallmann, J. Endophytic and ectophytic potato- associated bacterial communities differ in structure and antagonistic function against plant pathogenic fungi. FEMS Microbiol. Ecol. 2005, 51, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Faltin, F.; Lottmann, J.; Grosch, R.; Berg, G. Strategy to select and assess antagonistic bacteria for biological control of Rhizoctonia solani Kühn. Can. J. Microbiol. 2004, 50, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Scherwinski, K.; Grosch, R.; Berg, G. Effect of bacterial antagonists on lettuce: Active biocontrol of Rhizoctonia solani and negligible, short-term effects on nontarget microorganisms. FEMS Microbiol. Ecol. 2008, 64, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed]
- Krechel, A.; Faupel, A.; Hallmann, J.; Ulrich, A.; Berg, G. Potato-associated bacteria and their antagonistic potential towards plant-pathogenic fungi and the plant-parasitic nematode Meloidogyne incognita (Kofoid & White) Chitwood. Can. J. Microbiol. 2002, 48, 772–786. [Google Scholar] [PubMed]
- Madhaiyan, M.; Poonguzhali, S.; Saravanan, V.S.; Selvapravin, K.; Duraipandiyan, V.; Al-Dhabi, N.A. Pseudomonas sesami sp. nov., a plant growth-promoting Gammaproteobacteria isolated from the rhizosphere of Sesamum indicum L. Antonie van Leeuwenhoek 2017, 110, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.E.; Rossi-Tamisier, M.; Benamar, S.; Raoult, D. Cautionary tale of using 16S rRNA gene sequence similarity values in identification of human-associated bacterial species. Int. J. Syst. Evol. Microbiol. 2015, 65, 1929–1934. [Google Scholar]
- Tran, P.N.; Savka, M.A.; Gan, H.M. In-silico taxonomic classification of 373 genomes reveals species misidentification and new genospecies within the genus Pseudomonas. Front. Microbiol. 2017, 8, 1296. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Rodriguez-R, L.M.; Konstantinidis, K.T. MyTaxa: An advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res. 2014, 42, e73. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-R, L.M.; Konstantinidis, K.T. Bypassing Cultivation To Identify Bacterial Species. Microbe Mag. 2016, 9, 111–118. [Google Scholar] [CrossRef]
- Des Essarts, Y.R.; Cigna, J.; Quêtu-Laurent, A.; Caron, A.; Munier, E.; Beury-Cirou, A.; Hélias, V.; Faure, D. Biocontrol of the potato blackleg and soft rot diseases caused by Dickeya dianthicola. Appl. Environ. Microbiol. 2016, 82, 268–278. [Google Scholar] [CrossRef]
- Keel, C.; Weller, D.M.; Natsch, A.; Défago, G.; Cook, R.J.; Thomashow, L.S. Conservation of the 2,4-diacetylphloroglucinol biosynthesis locus among fluorescent Pseudomonas strains from diverse geographic locations. Appl. Environ. Microbiol. 1996, 62, 552–563. [Google Scholar]
- Eng, W.W.H.; Gan, H.M.; Gan, H.Y.; Hudson, A.O.; Savka, M.A. Whole-Genome Sequence and Annotation of Octopine-Utilizing Pseudomonas kilonensis (Previously P. fluorescens) Strain 1855-344. Genome Announc. 2015, 3, e00463-15. [Google Scholar] [CrossRef]
- Egelkamp, R.; Schneider, D.; Hertel, R.; Daniel, R. Nitrile-Degrading Bacteria Isolated from Compost. Front. Environ. Sci. 2017, 5, 56. [Google Scholar] [CrossRef]
- Blom, J.; Kreis, J.; Spänig, S.; Juhre, T.; Bertelli, C.; Ernst, C.; Goesmann, A. EDGAR 2.0: An enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 2016, 44, W22–W28. [Google Scholar] [CrossRef] [PubMed]
- Mitić, N.; Miraula, M.; Selleck, C.; Hadler, K.S.; Uribe, E.; Pedroso, M.M.; Schenk, G. Catalytic Mechanisms of Metallohydrolases Containing Two Metal Ions. Adv. Protein Chem. Struct. Biol. 2014, 97, 49–81. [Google Scholar] [PubMed]
- Jensen, S.O.; Reeves, P.R. Domain organisation in phosphomannose isomerases (types I and II). Biochim. Biophys. Acta 1998, 1382, 5–7. [Google Scholar] [CrossRef]
- Barry, S.M.; Challis, G.L. Recent advances in siderophore biosynthesis. Curr. Opin. Chem. Biol. 2009, 13, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Matelska, D.; Steczkiewicz, K.; Ginalski, K. Comprehensive classification of the PIN domain-like superfamily. Nucleic Acids Res. 2017, 45, 6995–7020. [Google Scholar] [CrossRef] [PubMed]
- Troxell, B.; Hassan, H.M. Transcriptional regulation by Ferric Uptake Regulator (Fur) in pathogenic bacteria. Front. Cell. Infect. Microbiol. 2013, 3, 59. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.M.; Grossman, A.D. Integrative and Conjugative Elements (ICEs): What They Do and How They Work. Annu. Rev. Genet. 2015, 49, 577–601. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, R.A.; Waldor, M.K. A toxin-antitoxin system promotes the maintenance of an integrative conjugative element. PLoS Genet. 2009, 5, e1000439. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Apweiler, R.; Alpi, E.; Antunes, R.; Arganiska, J.; Bely, B.; Bingley, M.; et al. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Hingamp, P.; Kojima, K.K.; Villar, E.; Romac, S.; Veluchamy, A.; Boccara, M.; Jaillon, O.; Iudicone, D.; Bowler, C.; et al. Reverse transcriptase genes are highly abundant and transcriptionally active in marine plankton assemblages. ISME J. 2016, 10, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Caparrós-Martín, J.A.; McCarthy-Suárez, I.; Culiáñez-Macià, F.A. HAD hydrolase function unveiled by substrate screening: enzymatic characterization of Arabidopsis thaliana subclass I phosphosugar phosphatase AtSgpp. Planta 2013, 237, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Favrot, L.; Blanchard, J.S.; Vergnolle, O. Bacterial GCN5-Related N-Acetyltransferases: From Resistance to Regulation. Biochemistry 2016, 55, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Loper, J.E.; Hassan, K.A.; Mavrodi, D.V.; Davis, E.W.; Lim, C.K.; Shaffer, B.T.; Elbourne, L.D.; Stockwell, V.O.; Hartney, S.L.; Breakwell, K.; et al. Comparative genomics of plant-associated pseudomonas spp.: Insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS Genet. 2012, 8, e1002784. [Google Scholar] [CrossRef] [PubMed]
- Van Der Voort, M.; Meijer, H.J.G.; Schmidt, Y.; Watrous, J.; Dekkers, E.; Mendes, R.; Dorrestein, P.C.; Gross, H.; Raaijmakers, J.M. Genome mining and metabolic profiling of the rhizosphere bacterium Pseudomonas sp. SH-C52 for antimicrobial compounds. Front. Microbiol. 2015, 6, 693. [Google Scholar] [CrossRef] [PubMed]
- Cimermancic, P.; Medema, M.H.; Claesen, J.; Kurita, K.; Wieland Brown, L.C.; Mavrommatis, K.; Pati, A.; Godfrey, P.A.; Koehrsen, M.; Clardy, J.; et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 2014, 158, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.P.; Hartmann, A.; Gao, X.W.; Borriss, R. Biocontrol mechanism by root-associated Bacillus amyloliquefaciens FZB42—A review. Front. Microbiol. 2015, 6, 780. [Google Scholar] [CrossRef]
- Doornbos, R.F.; Van Loon, L.C.; Bakker, P.A. Impact of root exudates and plant defense signaling on bacterial communities in the rhizosphere. A review. Agron. Sustain. Dev. 2012, 32, 227–243. [Google Scholar] [CrossRef]
- Venturi, V.; Keel, C. Signaling in the Rhizosphere. Trends Plant Sci. 2016, 21, 187–198. [Google Scholar] [CrossRef]
- Yi, H.S.; Ahn, Y.R.; Song, G.C.; Ghim, S.Y.; Lee, S.; Lee, G.; Ryu, C.M. Impact of a bacterial volatile 2,3-butanediol on Bacillus subtilis rhizosphere robustness. Front. Microbiol. 2016, 7, 993. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Jiang, W.; Cheng, Z.; Heikkila, J.J.; Glick, B.R. The complete genome sequence of the plant growth-promoting bacterium Pseudomonas sp. UW4. PLoS ONE 2013, 8, e58640. [Google Scholar] [CrossRef] [PubMed]
- Vitreschak, A.G. Regulation of riboflavin biosynthesis and transport genes in bacteria by transcriptional and translational attenuation. Nucleic Acids Res. 2002, 30, 3141–3151. [Google Scholar] [CrossRef] [PubMed]
- Dakora, F.D.; Matiru, V.N.; Kanu, A.S. Rhizosphere ecology of lumichrome and riboflavin, two bacterial signal molecules eliciting developmental changes in plants. Front. Plant Sci. 2015, 6, 700. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Marshall, N.C.; Rowland, J.L.; McCoy, J.M.; Worrall, L.J.; Santos, A.S.; Strynadka, N.C.; Finlay, B.B. Assembly, structure, function and regulation of type III secretion systems. Nat. Rev. Microbiol. 2017, 15, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.M.; Bertrand, N.; Rainey, P.B. Type III secretion in plant growth-promoting Pseudomonas fluorescens SBW25. Mol. Microbiol. 2001, 41, 999–1014. [Google Scholar] [CrossRef]
- Alori, E.T.; Glick, B.R.; Babalola, O.O. Microbial phosphorus solubilization and its potential for use in sustainable agriculture. Front. Microbiol. 2017, 8, 971. [Google Scholar] [CrossRef]
- Rodríguez, H.; Fraga, R.; Gonzalez, T.; Bashan, Y. Genetics of phosphate solubilization and its potential applications for improving plant growth-promoting bacteria. Plant Soil 2007, 287, 15–21. [Google Scholar] [CrossRef]
- Molina-Henares, M.A.; García-Salamanca, A.; Molina-Henares, A.J.; De La Torre, J.; Herrera, M.C.; Ramos, J.L.; Duque, E. Functional analysis of aromatic biosynthetic pathways in Pseudomonas putida KT2440. Microb. Biotechnol. 2009, 2, 91–100. [Google Scholar] [CrossRef]
- Lamb, A.L. Breaking a pathogen’s iron will: Inhibiting siderophore production as an antimicrobial strategy. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2015, 1854, 1054–1070. [Google Scholar] [CrossRef]
- Serino, L.; Reimmann, C.; Visca, P.; Beyeler, M.; Chiesa, V.D.; Haas, D. Biosynthesis of pyochelin and dihydroaeruginoic acid requires the iron-regulated pchDCBA operon in Pseudomonas aeruginosa. J. Bacteriol. 1997, 179, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Young, I.G.; Gibson, F. Regulation of the enzymes involved in the biosynthesis of 2,3-dihydroxybenzoic acid in Aerobacter aerogenes and Escherichia coli. BBA Gen. Subj. 1969, 177, 401–411. [Google Scholar] [CrossRef]
- Pupin, M.; Esmaeel, Q.; Flissi, A.; Dufresne, Y.; Jacques, P.; Leclère, V. Norine: A powerful resource for novel nonribosomal peptide discovery. Synth. Syst. Biotechnol. 2016, 1, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Mann, E.E.; Wozniak, D.J. Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol. Rev. 2012, 36, 893–916. [Google Scholar] [CrossRef]
- Malanovic, N.; Lohner, K. Gram-positive bacterial cell envelopes: The impact on the activity of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2016, 1858, 936–946. [Google Scholar] [CrossRef]
- Mellgren, E.M.; Kloek, A.P.; Kunkel, B.N. Mqo, a tricarboxylic acid cycle enzyme, is required for virulence of Pseudomonas syringae pv. tomato strain DC3000 on Arabidopsis thaliana. J. Bacteriol. 2009, 191, 3132–3141. [Google Scholar] [CrossRef]
- Neupane, S.; Finlay, R.D.; Alström, S.; Elfstrand, M.; Högberg, N. Transcriptional responses of the bacterial antagonist Serratia plymuthica to the fungal phytopathogen Rhizoctonia solani. Environ. Microbiol. Rep. 2015, 7, 123–127. [Google Scholar] [CrossRef]
- Perazzolli, M.; Herrero, N.; Sterck, L.; Lenzi, L.; Pellegrini, A.; Puopolo, G.; Van de Peer, Y.; Pertot, I. Transcriptomic responses of a simplified soil microcosm to a plant pathogen and its biocontrol agent reveal a complex reaction to harsh habitat. BMC Genom. 2016, 17, 838. [Google Scholar] [CrossRef]
- Achouak, W.; Conrod, S.; Cohen, V.; Heulin, T. Phenotypic Variation of Pseudomonas brassicacearum as a Plant Root-Colonization Strategy. Mol. Plant-Microbe Interact. 2004, 17, 872–879. [Google Scholar] [CrossRef]
- Van Den Broek, D.; Bloemberg, G.V.; Lugtenberg, B. The role of phenotypic variation in rhizosphere Pseudomonas bacteria. Environ. Microbiol. 2005, 7, 1686–1697. [Google Scholar] [CrossRef]
- Sajitha, K.L.; Dev, S.A. Quantification of antifungal lipopeptide gene expression levels in Bacillus subtilis B1 during antagonism against sapstain fungus on rubberwood. Biol. Control 2016, 96, 78–85. [Google Scholar] [CrossRef]
- Lu, X.; Zhou, D.; Chen, X.; Zhang, J.; Huang, H.; Wei, L. Isolation and characterization of Bacillus altitudinis JSCX-1 as a new potential biocontrol agent against Phytophthora sojae in soybean [Glycine max (L.) Merr.]. Plant Soil 2017, 416, 53–66. [Google Scholar] [CrossRef]
- Blauth de Lima, F.; da Silva Ribeiro, R.T.; Félix, C.; Osório, N.; Alves, A.; Esteves, A.C.; Vitorino, R.; Domingues, P. Trichoderma harzianum T1A constitutively secretes proteins involved in the biological control of Guignardia citricarpa. Biol. Control 2017, 106, 99–109. [Google Scholar] [CrossRef]
- Puehringer, S.; Metlitzky, M.; Schwarzenbacher, R. The pyrroloquinoline quinone biosynthesis pathway revisited: A structural approach. BMC Biochem. 2008, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Han, S.H.; Kim, C.H.; Lee, J.H.; Park, J.Y.; Cho, S.M.; Park, S.K.; Kim, K.Y.; Krishnan, H.B.; Kim, Y.C. Inactivation of pqq genes of Enterobacter intermedium 60-2G reduces antifungal activity and induction of systemic resistance. FEMS Microbiol. Lett. 2008, 282, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Hernández, V.M.; Girard, L.; Hernández-Lucas, I.; Vázquez, A.; Ortíz-Ortíz, C.; Díaz, R.; Dunn, M.F. Genetic and biochemical characterization of arginine biosynthesis in Sinorhizobium meliloti 1021. Microbiology 2015, 161, 1671–1682. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zúñiga, M.; Pérez, G.; González-Candelas, F. Evolution of arginine deiminase (ADI) pathway genes. Mol. Phylogenetics Evol. 2002, 25, 429–444. [Google Scholar] [CrossRef]
- Pitcher, R.S.; Watmough, N.J. The bacterial cytochrome cbb3 oxidases. Biochim. Biophys. Acta 2004, 1655, 388–399. [Google Scholar] [CrossRef]
- Hilker, R.; Stadermann, K.B.; Schwengers, O.; Anisiforov, E.; Jaenicke, S.; Weisshaar, B.; Zimmermann, T.; Goesmann, A. ReadXplorer 2—Detailed read mapping analysis and visualization from one single source. Bioinformatics 2016, 32, 3702–3708. [Google Scholar] [CrossRef]
- Krishna, S.S.; Aravind, L.; Bakolitsa, C.; Caruthers, J.; Carlton, D.; Miller, M.D.; Abdubek, P.; Astakhova, T.; Axelrod, H.L.; Chiu, H.J.; et al. The structure of SSO2064, the first representative of Pfam family PF01796, reveals a novel two-domain zinc-ribbon OB-fold architecture with a potential acyl-CoA-binding role. Acta Crystallogr. Sect. F Struct. Biol. Crystall. Commun. 2010, 66, 1160–1166. [Google Scholar] [CrossRef]
- Zhang, L.; Xu, Q.; Zhan, S.; Li, Y.; Lin, H.; Sun, S.; Sha, L.; Hu, K.; Guan, X.; Shen, Y. A new NAD(H)-dependent meso-2,3-butanediol dehydrogenase from an industrially potential strain Serratia marcescens H30. Appl. Microbiol. Biotechnol. 2014, 98, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Ryu, C.M.; Farag, M.A.; Hu, C.H.; Reddy, M.S.; Wei, H.X.; Pare, P.W.; Kloepper, J.W. Bacterial volatiles promote growth in Arabidopsis. Proc. Natl. Acad. Sci. USA 2003, 100, 4927–4932. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Preciado, A.; Torres, A.G.; Merino, E.; Bonomi, H.R.; Goldbaum, F.A.; García-Angulo, V.A. Extensive identification of bacterial riboflavin transporters and their distribution across bacterial species. PLoS ONE 2015, 10, e0126124. [Google Scholar] [CrossRef] [PubMed]
- García-Angulo, V.A. Overlapping riboflavin supply pathways in bacteria. Crit. Rev. Microbiol. 2017, 43, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Tatusova, T.; Dicuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Van Heel, A.J.; de Jong, A.; Montalbán-López, M.; Kok, J.; Kuipers, O.P. BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 2013, 41, W448–W453. [Google Scholar] [CrossRef]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Müller, R.; Wohlleben, W.; et al. AntiSMASH 3.0-A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef]
- Flissi, A.; Dufresne, Y.; Michalik, J.; Tonon, L.; Janot, S.; Noé, L.; Jacques, P.; Leclére, V.; Pupin, M. Norine, the knowledgebase dedicated to non-ribosomal peptides, is now open to crowdsourcing. Nucleic Acids Res. 2016, 44, D1113–D1118. [Google Scholar] [CrossRef]
- Sneath, P.H.A.; McGowan, V.; Skerman, V.B.D. Approved lists of bacterial names. Med. J. Aust. 1980, 2, 3–4. [Google Scholar]
- Elomari, M.; Coroler, L.; Hoste, B.; Gillis, M.; Izard, D.; Leclerc, H. DNA Relatedness among Pseudomonas Strains Isolated from Natural Mineral Waters and Proposal of Pseudomonas veronii sp. nov. Int. J. Syst. Bacteriol. 2009, 46, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, U.; Ulrich, A.; Schumann, P. Fluorescent pseudomonads associated with the phyllosphere of grasses; Pseudomonas trivialis sp. nov., Pseudomonas poae sp. nov. and Pseudomonas congelans sp. nov. Int. J. Syst. Evol. Microbiol. 2003, 53, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Euzéby, J. Validation of publication of new names and new combinations previously effectively published outside the USEM. Int. J. Syst. Evol. Microbiol. 2004, 54, 631–632. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhang, H.; Gao, S.; Lercher, M.J.; Chen, W.H.; Hu, S. Evolview v2: An online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 2016, 44, W236–W241. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Grosch, R.; Schneider, J.H.; Kofoet, A. Characterisation of Rhizoctonia solani anastomosis groups causing bottom rot in field-grown lettuce in Germany. Eur. J. Plant Pathol. 2004, 110, 53–62. [Google Scholar] [CrossRef]
- Pátek, M.; Holátko, J.; Busche, T.; Kalinowski, J.; Nešvera, J. Corynebacterium glutamicum promoters: A practical approach. Microb. Biotechnol. 2013, 6, 103–117. [Google Scholar] [CrossRef]
- Haas, B.J.; Chin, M.; Nusbaum, C.; Birren, B.W.; Livny, J. How deep is deep enough for RNA-Seq profiling of bacterial transcriptomes? BMC Genom. 2012, 13, 734. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware AlignerLawrence Berkeley National Lab.(LBNL), Berkeley, CA (United States). 2014. Available online: https://jgi.doe.gov/wp-content/uploads/2013/11/BB_User-Meeting-2014-poster-FINAL.pdf (accessed on 7 August 2019).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism/Name | Strain | Size [Mb] | GC [%] | Scaf-Folds | Genes | Niche/Source | References | AAI |
|---|---|---|---|---|---|---|---|---|
| P. brassicacearum | 3Re2-7 | 6.7 | 60.8 | 1 | 6267 | Potato endorhiza | [32], this study | 99.9 |
| P. brassicacearum subsp. brassicacearum | NFM421 | 6.8 | 60.8 | 1 | 6209 | Arabidopsis thaliana rhizoplane | [16,18] | 100 |
| P. brassicacearum | DF41 | 6.6 | 60.5 | 1 | 5884 | Canola root | [19] | 96 |
| P. brassicacearum | LBUM300 | 6.9 | 60.8 | 1 | 6308 | Strawberry rhizosphere | [22] | 99.9 |
| P. brassicacearum | L13-6-12 | 6.7 | 60.9 | 1 | 6046 | Potato rhizosphere | [21] | 99.9 |
| P. brassicacearum | BS3663 | 6.7 | 60.8 | 1 | 6129 | No metadata available | Unpublished | 99.9 |
| P. brassicacearum | 51MFCVI2.1 | 6.5 | 61.0 | 49 | 5980 | Rhizosphere | Unpublished | 99.9 |
| P. brassicacearum | PP1_210F | 6.7 | 60.8 | 5 | 6159 | Peels tuber | [20,38] | 99.9 |
| P. brassicacearum | PA1G7 | 6.7 | 60.8 | 8 | 6180 | Potato field soil | [20,38] | 99.9 |
| P. brassicacearum | LZ-4 | 6.2 | 60.1 | 111 | 5642 | Yellow River | Unpublished | 94.9 |
| P. brassicacearum | TM1A3 | 6.6 | 60.9 | 29 | 6037 | Tomato rhizosphere | [39] | 99.8 |
| P. brassicacearum | Wood3 | 6.3 | 62.3 | 433 | 5752 | Agricultural | Unpublished | 94.7 |
| P. brassicacearum | 36B7 | 7.1 | 60.7 | 28 | 6411 | Agricultural | Unpublished | 95.8 |
| P. brassicacearum | 36D4 | 7.0 | 60.5 | 128 | 6290 | Agricultural | Unpublished | 95.8 |
| P. brassicacearum | Delaware | 6.8 | 60.8 | 226 | 6263 | Agricultural | Unpublished | 99.8 |
| P. brassicacearum | 93F8 | 6.8 | 61.0 | 36 | 6291 | Agricultural | Unpublished | 99.9 |
| P. brassicacearum | 37D10 | 6.3 | 58.8 | 55 | 5946 | Agricultural | Unpublished | 88.6 |
| P. brassicacearum | Wood1 | 6.9 | 60.8 | 187 | 6288 | Agricultural | Unpublished | 99.8 |
| P. brassicacearum | 48H11 | 6.0 | 58.8 | 36 | 5510 | Agricultural | Unpublished | 88.7 |
| P. brassicacearum | 38D7 | 6.5 | 59.5 | 485 | 6087 | Agricultural | Unpublished | 89.2 |
| P. brassicacearum | 38D4 | 7.1 | 58.7 | 199 | 6793 | Agricultural | Unpublished | 89.5 |
| P. kilonensis | 1855-344 | 6.8 | 60.7 | 73 | 6146 | Soil | [40] | 98.2 |
| P. kilonensis | P12 | 6.4 | 60.8 | 44 | 5791 | Tobacco | [39] | 98.2 |
| P. kilonensis | DSM 13647 | 6.4 | 60.9 | 44 | 5758 | No metadata available | Unpublished | 98.1 |
| P. kilonensis | ACN7 | 6.5 | 61 | 200 | 5897 | Compost soil | [41] | 98.2 |
| P. kilonensis | ACN4 | 6.5 | 60.8 | 91 | 5956 | Compost soil | [41] | 98.2 |
| P. kilonensis | ZKA7 | 6.8 | 60.6 | 1 | 6158 | No metadata available | Unpublished | 97.9 |
| P. kilonensis | BS3780 | 6.4 | 60.8 | 2 | 5775 | No metadata available | Unpublished | 98.1 |
| Locus tag | Predicted Function |
|---|---|
| ELZ14_04045 | hypothetical protein |
| ELZ14_04050 | DNA-binding protein |
| ELZ14_04055 | hypothetical protein |
| ELZ14_04060 | hypothetical protein |
| ELZ14_04065 | hypothetical protein |
| ELZ14_04070 | hypothetical protein |
| ELZ14_04075 | nucleotidyltransferase family protein |
| ELZ14_04080 * | hypothetical protein |
| ELZ14_04085 * | MafI family immunity protein |
| ELZ14_04090 | hypothetical protein |
| ELZ14_04095 | hypothetical protein |
| ELZ14_04100 | hypothetical protein |
| ELZ14_04105 | DUF4935 domain-containing protein |
| ELZ14_04110 | hypothetical protein |
| ELZ14_04115 * | hypothetical protein |
| ELZ14_04120 | hypothetical protein |
| ELZ14_04125 * | hypothetical protein |
| ELZ14_04130 * | hypothetical protein |
| ELZ14_04135 * | integrase |
| ELZ14_04140 | hypothetical protein |
| ELZ14_04145 | hypothetical protein |
| ELZ14_04150 | hypothetical protein |
| ELZ14_04155 | hypothetical protein |
| ELZ14_04160 | metallohydrolase |
| Locus Tag | Predicted Function |
|---|---|
| ELZ14_15030 | hypothetical protein |
| ELZ14_15035 * | hypothetical protein |
| ELZ14_15040 | helicase IV |
| ELZ14_15045 * | HNH endonuclease |
| ELZ14_15050 | hypothetical protein |
| ELZ14_15055 | endonuclease |
| ELZ14_15060 | hypothetical protein |
| ELZ14_15065 * | hypothetical protein |
| ELZ14_15070 | hypothetical protein |
| ELZ14_15075 | hypothetical protein |
| ELZ14_15080 | hypothetical protein |
| ELZ14_15085 | CHAT domain-containing protein |
| ELZ14_15090 | hypothetical protein |
| ELZ14_15095 * | DNA/RNA non-specific endonuclease |
| ELZ14_15100 * | hypothetical protein |
| ELZ14_15105 * | relaxase |
| ELZ14_15110 | DUF3742 family protein |
| ELZ14_15115 * | conjugal transfer protein TraG |
| ELZ14_15120 * | integrating conjugative element protein |
| ELZ14_15125 * | TIGR03756 family integrating conjugative element protein |
| ELZ14_15130 * | DNA repair protein RadC |
| ELZ14_15135 | hypothetical protein |
| (...) 32 * | |
| ELZ14_15295 | HAD family hydrolase |
| ELZ14_15300 | DNA-processing protein DprA |
| ELZ14_15305 * | hypothetical protein |
| ELZ14_15310 | hypothetical protein |
| ELZ14_15315 | DUF4935 domain-containing protein |
| ELZ14_15320 | hypothetical protein |
| ELZ14_15325 | DUF3800 domain-containing protein |
| ELZ14_15330 | toll/interleukin-1 receptor domain-containing protein |
| ELZ14_15335 | RNA-directed DNA polymerase |
| ELZ14_15340 | hypothetical protein |
| ELZ14_15345 * | hypothetical protein |
| ELZ14_15350 * | transposase |
| ELZ14_15355 | hypothetical protein |
| ELZ14_15360 * | hypothetical protein |
| ELZ14_15365 | hypothetical protein |
| ELZ14_15370 * | hypothetical protein |
| ELZ14_15375 | ATP-binding protein |
| ELZ14_15380 * | hypothetical protein |
| ELZ14_15385 * | leucine ABC transporter subunit substrate-binding protein LivK |
| ELZ14_15390 | hypothetical protein |
| Locus Tag | Predicted Function |
|---|---|
| ELZ14_26355 | hypothetical protein |
| ELZ14_26360 | hypothetical protein |
| ELZ14_26365 | DNA helicase |
| ELZ14_26370 | GNAT family N-acetyltransferase |
| Biocontrol Trait | Locus Tag (Gene Name) | Predicted EC Number and Protein Function | log2(TPM ) |
|---|---|---|---|
| Secondary metabolism & antibiotics | |||
| ELZ14_11930 (phlE) | n.a.; MFS transporter | 8.18 | |
| ELZ14_11935 (phlD) | EC 2.3.1.-; type III polyketide synthase | 11.05 | |
| ELZ14_11940 (phlB) | n.a.; 2,4-diacetylphloroglucinol biosynthesis protein | 10.12 | |
| DAPG synthesis | ELZ14_11945 (phlC) | n.a.; thiolase family protein | 10.79 |
| ELZ14_11950 (phlA) | EC 2.3.3.10; hydroxymethylglutaryl-CoA synthase | 10.86 | |
| ELZ14_11955 (phlF) | n.a.; TetR/AcrR family transcriptional regulator | 5.23 | |
| ELZ14_11960 (phlG) | n.a.; 2,4-diacetylphloroglucinol hydrolase | 10.97 | |
| ELZ14_11965 (phlH) | n.a.; TetR/AcrR family transcriptional regulator | 7.07 | |
| ELZ14_17910 (hcnC) | EC 1.4.99.5; cyanide-forming glycine dehydrogenase subunit HcnC | 9.33 | |
| HCN synthesis | ELZ14_17915 (hcnB) | EC 1.4.99.5; cyanide-forming glycine dehydrogenase subunit HcnB | 8.95 |
| ELZ14_17920 (hcnA) | EC 1.4.99.5; cyanide-forming glycine dehydrogenase subunit HcnA | 9.89 | |
| Biocontrol Trait | Locus Tag (Gene Name) | Predicted EC Number and Protein Function | log2 (TPM ) |
|---|---|---|---|
| ELZ14_17085 (-) | EC 1.1.1.4; 2,3-butanediol dehydrogenase | 4.47 | |
| ELZ14_17090 (acoC) | EC 2.3.1.12; acetoin dehydrogenase dihydrolipoyllysine-residue acetyltransferase subunit | 4.51 | |
| ELZ14_17095 (acoB) | EC 1.1.1.-; alpha-ketoacid dehydrogenase subunit beta | 4.55 | |
| Volatiles | ELZ14_17100 (acoA) | EC 1.1.1.-; thiamine pyrophosphate-dependent dehydrogenase E1 component subunit alpha | 4.58 |
| ELZ14_26505 (ilvC) | EC 1.1.1.86; Ketol-acid reductoisomerase | 8.25 | |
| ELZ14_26510 (ilvH) | EC 2.2.1.6; Acetolactate synthase isozyme 3 small subunit | 6.78 | |
| ELZ14_26515 (ilvI) | EC 2.2.1.6; Acetolactate synthase isozyme 3 large subunit | 6.13 | |
| ELZ14_12055 (ilvX) | EC 2.2.1.6; Putative acetolactate synthase large subunit IlvX | 1.81 | |
| ELZ14_11285 (acdS) | EC 3.5.99.7; 1-aminocyclopropane-1-carboxylate deaminase | 4.66 | |
| ELZ14_14325 (ribBA) | n.a.; 3,4-dihydroxy-2-butanone-4-phosphate synthase/GTP cyclohydrolase II | 1.98 | |
| ELZ14_23780 (ribH) | EC 2.5.1.78; 6,7-dimethyl-8-ribityllumazine synthase | 4.92 | |
| ELZ14_26870 (ribF) | EC 2.7.1.26; bifunctional riboflavin kinase/FAD synthetase | 5.40 | |
| ELZ14_27845 (ribA) | EC 3.5.4.25; GTP cyclohydrolase II | 5.63 | |
| ELZ14_27870 (ribH2) | EC 2.5.1.78; 6,7-dimethyl-8-ribityllumazine synthase | 8.05 | |
| Plant growth promotion | ELZ14_27875 (ribB) | n.a.; 3,4-dihydroxy-2-butanone-4-phosphate synthase | 4.82 |
| ELZ14_27880 (ribE) | EC 2.5.1.9; riboflavin synthase | 4.69 | |
| ELZ14_27885 (ribD) | n.a.; riboflavin biosynthesis protein RibD | 3.80 | |
| ELZ14_28390 (trpC) | EC 4.1.1.48; indole-3-glycerol phosphate synthase TrpC | 6.23 | |
| ELZ14_28395 (trpD) | EC 2.4.2.18; anthranilate phosphoribosyltransferase | 5.98 | |
| ELZ14_28400 (trpG) | EC 4.1.3.27; aminodeoxychorismate/anthranilate synthase component II | 5.02 | |
| ELZ14_28405 (estA) | EC 3.1.1.1; autotransporter domain-containing esterase | 3.90 | |
| ELZ14_28410 (trpE) | EC 4.1.3.27; anthranilate synthase component I | 4.43 | |
| ELZ14_15680 (phyC) | EC 3.1.3.8; phytase | 0.18 | |
| ELZ14_27855 (pgpA) | EC 3.1.3.27; phosphatidylglycerophosphatase A | 6.49 | |
| ELZ14_28415 (gph) | EC 3.1.3.18; phosphoglycolate phosphatase | 4.60 | |
| ELZ14_28615 (pqqF) | EC:3.4.24.-; pyrroloquinoline quinone biosynthesis protein PqqF | 3.98 | |
| ELZ14_28620 (pqqA) | n.a.; pyrroloquinoline quinone precursor peptide PqqA | 12.77 | |
| ELZ14_28625 (pqqB) | n.a.; pyrroloquinoline quinone biosynthesis protein PqqB | 6.86 | |
| ELZ14_28630 (pqqC) | EC 1.3.3.11; pyrroloquinoline-quinone synthase PqqC | 5.99 | |
| Phosphate solubilization | ELZ14_28635 (pqqD) | n.a.; pyrroloquinoline quinone biosynthesis peptide chaperone PqqD | 6.32 |
| ELZ14_28640 (pqqE) | n.a.; pyrroloquinoline quinone biosynthesis protein PqqE | 5.96 | |
| ELZ14_30735 (-) | EC 3.1.3.16; serine/threonine-protein phosphatase | 5.10 | |
| ELZ14_08470 (mupP) | EC 3.1.3.105; N-acetylmuramic acid 6-phosphate phosphatase MupP | 7.05 | |
| ELZ14_08475 (ubiG) | EC 2.1.1.222; 3-demethylubiquinol 3-O-methyltransferase UbiG | 7.36 | |
| ELZ14_04440 (-) | EC 3.1.3.1; alkaline phosphatase | 6.63 | |
| ELZ14_04880 (-) | EC 3.1.3.1; alkaline phosphatase family protein | 2.57 | |
| ELZ14_09815 (-) | EC 3.1.3.16; phosphoprotein phosphatase | 3.15 |
| Biocontrol Trait | Locus Tag (Gene Name) | Predicted EC Number and Protein Function | log2 (TPM ) |
|---|---|---|---|
| ELZ14_01095 (-) | n.a.; TonB-dependent siderophore receptor | 0.32 | |
| ELZ14_04245 (fur) | n.a.; ferric iron uptake transcriptional regulator | 8.94 | |
| ELZ14_10605 (-) | n.a.; TonB-dependent siderophore receptor | 1.14 | |
| ELZ14_12290 (hcsK) | n.a.; siderophore-iron reductase FhuF | 4.09 | |
| ELZ14_13330 (pchB) | n.a.; isochorismate lyase | 4.10 | |
| ELZ14_14280 (-) | n.a.; TonB-dependent siderophore receptor | 2.35 | |
| ELZ14_16995 (-) | n.a.; TonB-dependent siderophore receptor | 1.85 | |
| Iron acquisition | ELZ14_18660 (-) | n.a.; TonB-dependent siderophore receptor | 0.84 |
| ELZ14_21210 (pvdA) | EC 1.14.13.196; ornithine monooxygenase | 1.30 | |
| ELZ14_21260 (-) | n.a.; nonribosomal peptide synthetase | 1.42 | |
| ELZ14_21280 (-) | n.a.; TonB-dependent siderophore receptor | 2.82 | |
| ELZ14_21285 (-) | n.a.; nonribosomal peptide synthetase | 3.41 | |
| ELZ14_24950 (-) | n.a.; siderophore-interacting protein | 4.42 | |
| ELZ14_26060 (-) | n.a.; TonB-dependent siderophore receptor | 0.26 | |
| ELZ14_28775 (-) | n.a.; TonB-dependent siderophore receptor | 0.46 | |
| ELZ14_14600 (-) | n.a.; matrixin family metalloprotease | 12.59 | |
| ELZ14_14610 (prsD) | n.a.; type I secretion system permease/ATPase PrsD | 5.81 | |
| Exoprotease activity | ELZ14_14615 (-) | n.a.; HlyD family type I secretion periplasmic adaptor subunit | 6.32 |
| ELZ14_14620 (-) | n.a.; peptidase | 4.73 | |
| ELZ14_20130 (-) | n.a.; Hcp family type VI secretion system effector | 1.77 | |
| Chitinase activity | ELZ14_25035 (nagA) | EC 3.5.1.25; N-acetylglucosamine-6-phosphate deacetylase | 2.72 |
| Biocontrol Trait | Locus Tag (Gene Name) | Predicted EC Number and Protein Function | log2 (TPM ) |
|---|---|---|---|
| ELZ14_02400 (gltB) | EC 1.4.1.13; glutamate synthase large subunit | 5.12 | |
| ELZ14_02405 (gltD1) | EC 1.4.1.13; glutamate synthase small subunit | 5.59 | |
| ELZ14_02380 (aroK) | EC 2.7.1.71; shikimate kinase AroK | 5.44 | |
| Metabolism | ELZ14_02385 (aroB) | EC 4.2.3.4; 3-dehydroquinate synthase | 5.04 |
| ELZ14_03505 (aroQ) | EC 4.2.1.10; type II 3-dehydroquinate dehydratase | 5.88 | |
| ELZ14_08510 (aroA) | EC 2.5.1.19; 3-phosphoshikimate 1-carboxyvinyltransferase | 6.40 | |
| ELZ14_11900 (-) | n.a.; polyketide cyclase | 2.67 | |
| ELZ14_25290 (algD) | n.a.; nucleotide sugar dehydrogenase | 9.65 | |
| ELZ14_25300 (alg44) | n.a.; alginate biosynthesis protein Alg44 | 7.31 | |
| ELZ14_25320 (algX) | n.a.; alginate O-acetyltransferase | 7.49 | |
| ELZ14_25325 (algL) | EC 4.2.2.3; mannuronate-specific alginate lyase | 7.89 | |
| Exopolysaccharides | ELZ14_25335 (algJ) | n.a.; alginate O-acetyltransferase | 7.22 |
| ELZ14_25340 (algF) | n.a.; alginate O-acetyltransferase | 8.93 | |
| ELZ14_25350 (algA) | n.a.; alginate biosynthesis protein AlgA | 10.73 | |
| ELZ14_27635 (sacB) | EC 2.4.1.10; glycoside hydrolase 68 family protein (levansucrase) | 6.33 | |
| ELZ14_00175 (katE) | EC 1.11.1.6; catalase HPII | 8.02 | |
| ELZ14_03625 (copA) | EC 3.6.3.54; copper-exporting P-type ATPase A | 3.70 | |
| ELZ14_03915 (katG) | EC 1.11.1.21; catalase/peroxidase HPI | 6.31 | |
| ELZ14_09480 (-) | EC 3.6.3.54; cadmium-translocating P-type ATPase | 5.50 | |
| Detoxification | ELZ14_24395 (copA) | n.a.; copper resistance system multicopper oxidase | 2.25 |
| ELZ14_24400 (copB) | n.a.; copper resistance protein B | 1.40 | |
| ELZ14_24405 (copC) | n.a.; Copper resistance protein C | 3.03 | |
| ELZ14_24410 (copD) | n.a.; Copper resistance D family protein | 2.07 | |
| ELZ14_24510 (dps2) | EC 1.16.-.-; DNA starvation/stationary phase protection protein | 9.59 | |
| ELZ14_27055 (katB) | EC 1.11.1.6; catalase | 2.63 | |
| Lipopolysaccharides | ELZ14_31170 (-) | n.a.; lipoteichoic acid (LTA) synthase family protein | 1.48 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelkner, J.; Torres Tejerizo, G.; Hassa, J.; Lin, T.W.; Witte, J.; Verwaaijen, B.; Winkler, A.; Bunk, B.; Spröer, C.; Overmann, J.; et al. Genetic Potential of the Biocontrol Agent Pseudomonas brassicacearum (Formerly P. trivialis) 3Re2-7 Unraveled by Genome Sequencing and Mining, Comparative Genomics and Transcriptomics. Genes 2019, 10, 601. https://doi.org/10.3390/genes10080601
Nelkner J, Torres Tejerizo G, Hassa J, Lin TW, Witte J, Verwaaijen B, Winkler A, Bunk B, Spröer C, Overmann J, et al. Genetic Potential of the Biocontrol Agent Pseudomonas brassicacearum (Formerly P. trivialis) 3Re2-7 Unraveled by Genome Sequencing and Mining, Comparative Genomics and Transcriptomics. Genes. 2019; 10(8):601. https://doi.org/10.3390/genes10080601
Chicago/Turabian StyleNelkner, Johanna, Gonzalo Torres Tejerizo, Julia Hassa, Timo Wentong Lin, Julian Witte, Bart Verwaaijen, Anika Winkler, Boyke Bunk, Cathrin Spröer, Jörg Overmann, and et al. 2019. "Genetic Potential of the Biocontrol Agent Pseudomonas brassicacearum (Formerly P. trivialis) 3Re2-7 Unraveled by Genome Sequencing and Mining, Comparative Genomics and Transcriptomics" Genes 10, no. 8: 601. https://doi.org/10.3390/genes10080601
APA StyleNelkner, J., Torres Tejerizo, G., Hassa, J., Lin, T. W., Witte, J., Verwaaijen, B., Winkler, A., Bunk, B., Spröer, C., Overmann, J., Grosch, R., Pühler, A., & Schlüter, A. (2019). Genetic Potential of the Biocontrol Agent Pseudomonas brassicacearum (Formerly P. trivialis) 3Re2-7 Unraveled by Genome Sequencing and Mining, Comparative Genomics and Transcriptomics. Genes, 10(8), 601. https://doi.org/10.3390/genes10080601