Identification and Validation of Novel Reference Genes in Acute Lymphoblastic Leukemia for Droplet Digital PCR

 ,
,  and
and
Abstract
1. Introduction
2. Materials and methods
2.1. Gene Expression Data Mining
2.2. Tumor Samples
2.3. RNA Extraction
2.4. cDNA Synthesis
2.5. RT-PCR Amplification
2.6. Quantitative RT-PCR
2.7. Droplet Digital PCR
2.8. Statistical Analysis
3. Results
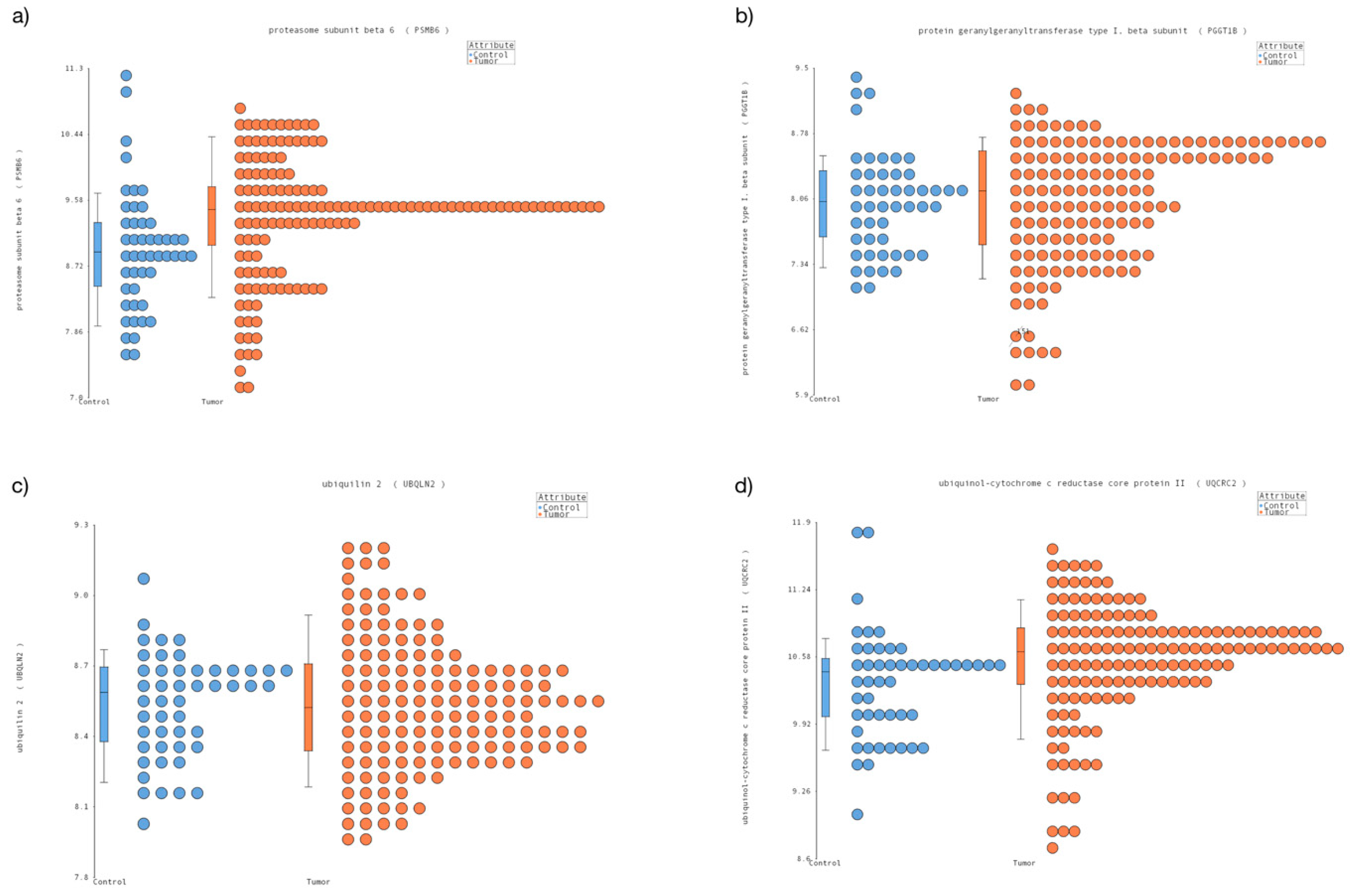
3.1. New Candidates Reference Genes in Pediatric Cancer
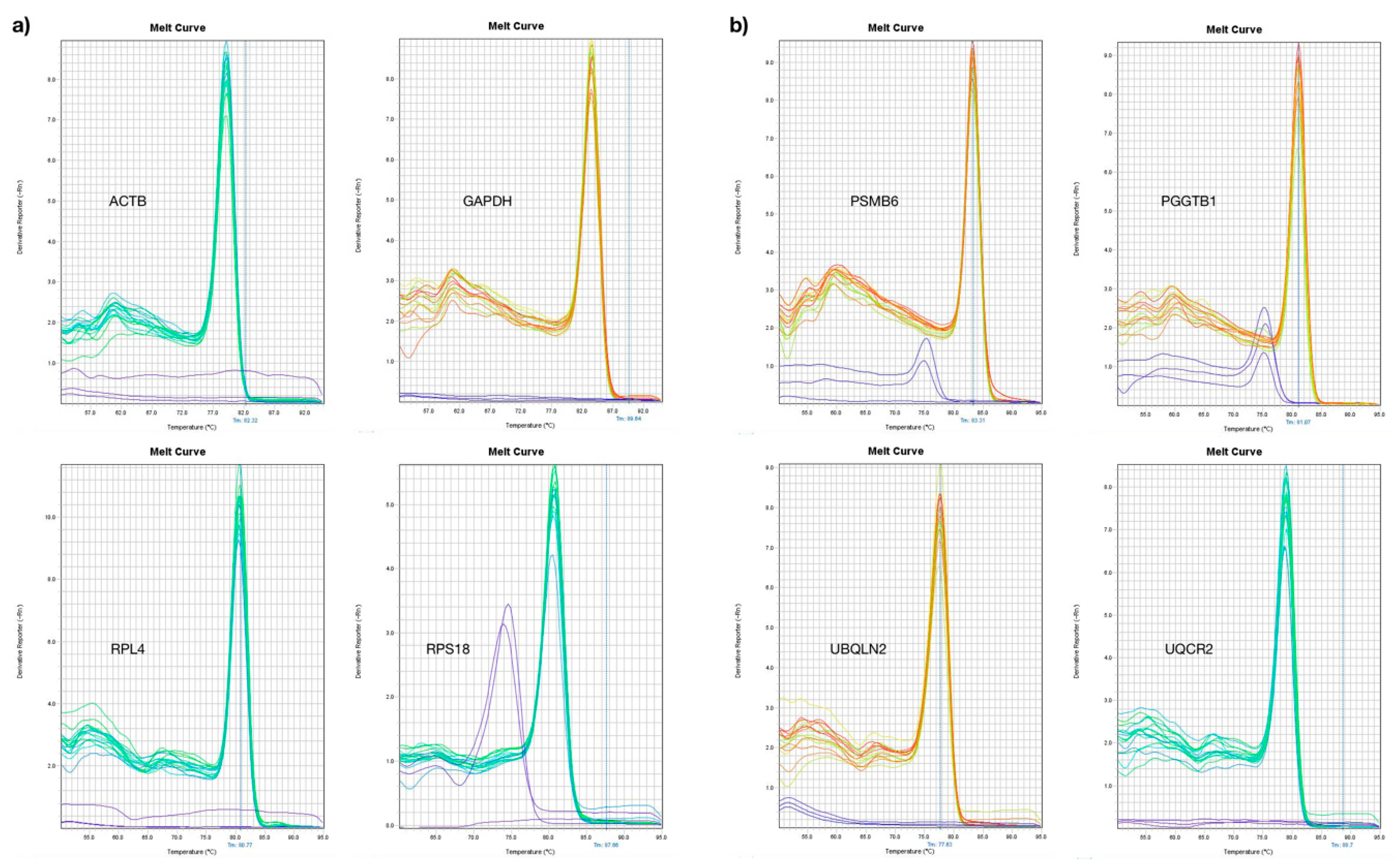
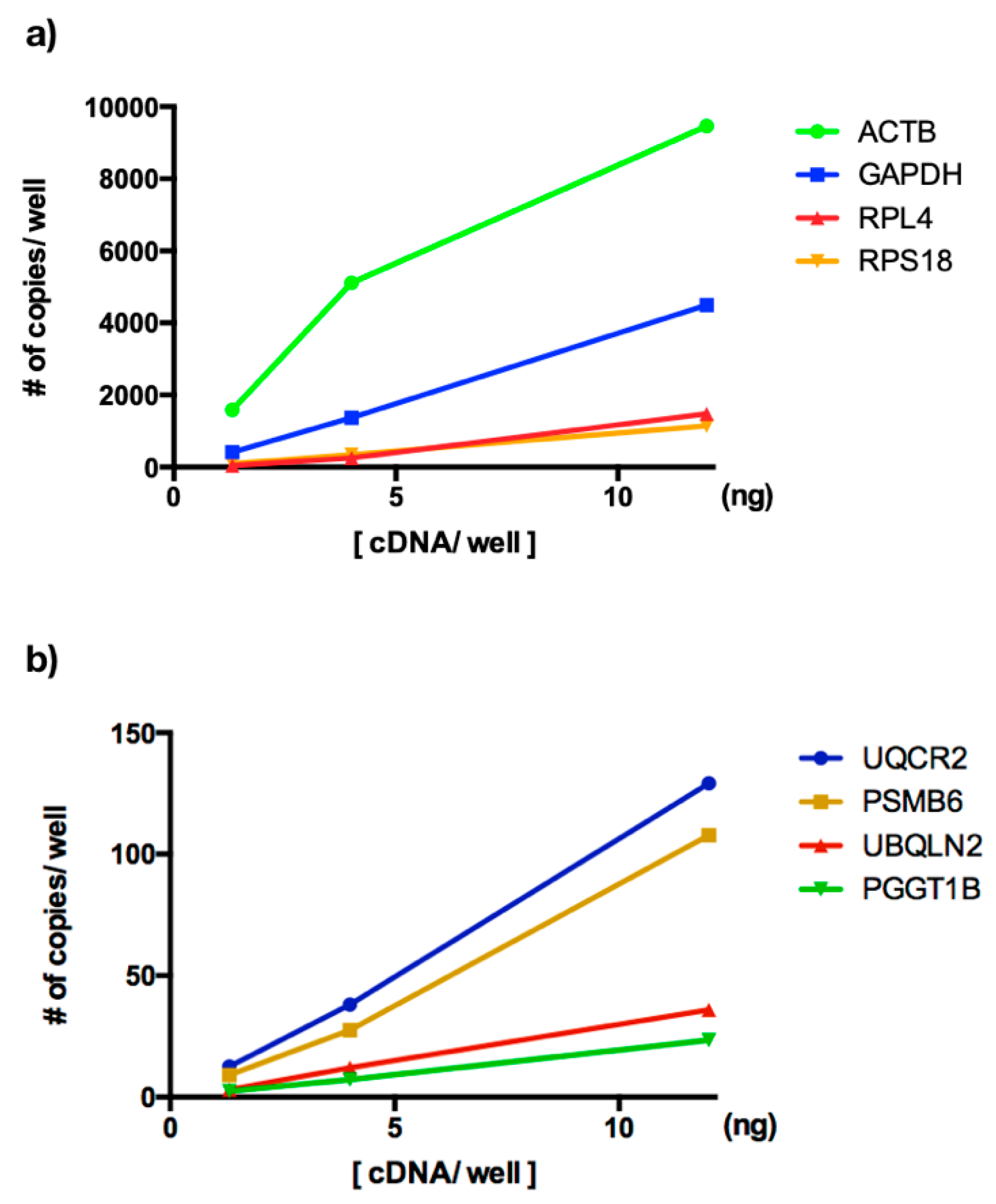
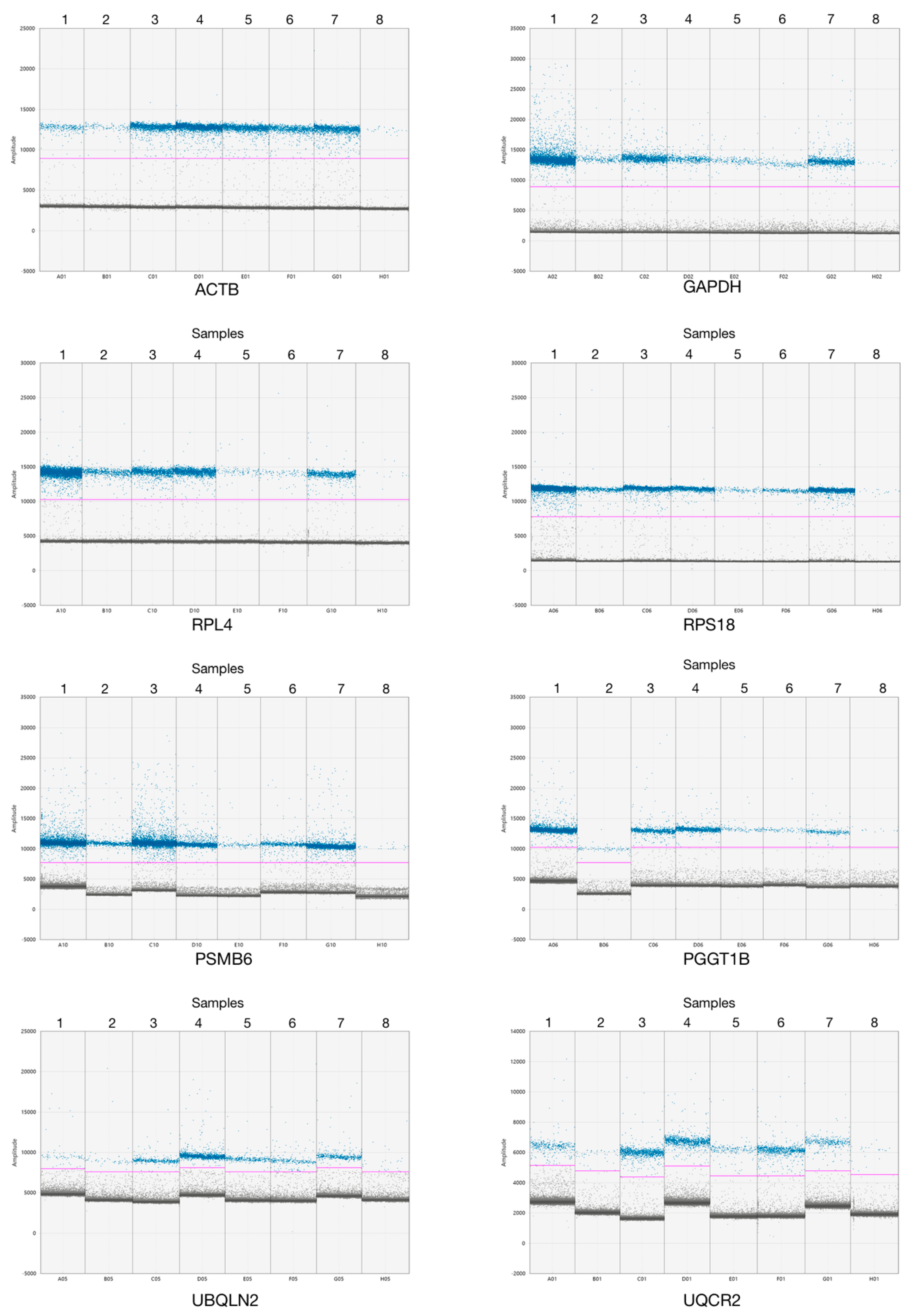
3.2. ddPCR Assay Optimization for Analysis of New Reference Genes
3.3. Reference Genes Validation in Acute Lymphoblastic Leukemia
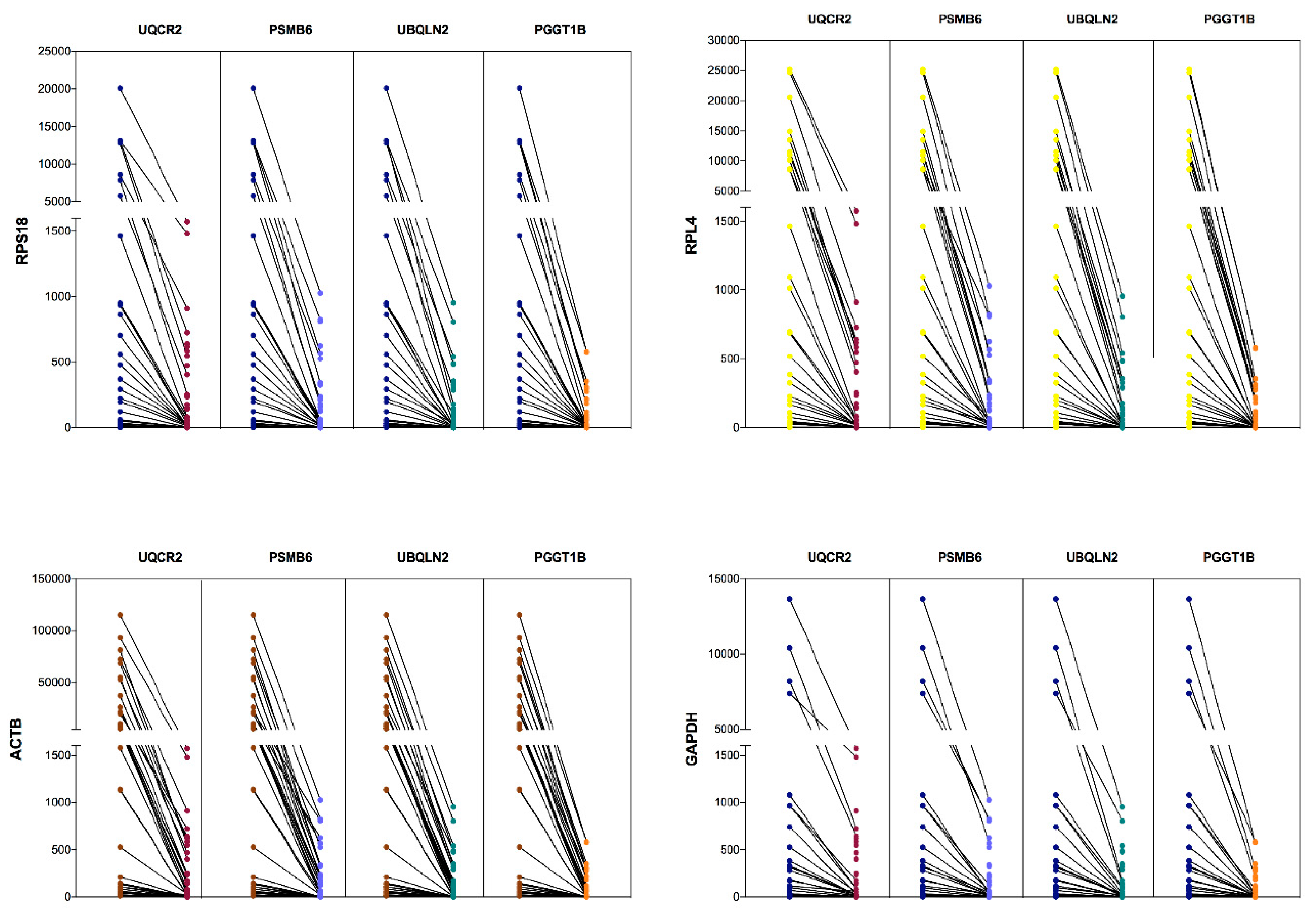
3.4. Comparative Expression Between CRGs and NRGs in Leukemia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Perez-Saldivar, M.L.; Fajardo-Gutierrez, A.; Bernaldez-Rios, R.; Martinez-Avalos, A.; Medina-Sanson, A.; Espinosa-Hernandez, L.; Flores-Chapa Jde, D.; Amador-Sanchez, R.; Penaloza-Gonzalez, J.G.; Alvarez-Rodriguez, F.J.; et al. Childhood acute leukemias are frequent in Mexico city: Descriptive epidemiology. BMC Cancer 2011, 11, 355. [Google Scholar] [CrossRef]
- Pritchard-Jones, K.; Pieters, R.; Reaman, G.H.; Hjorth, L.; Downie, P.; Calaminus, G.; Naafs-Wilstra, M.C.; Steliarova-Foucher, E. Sustaining innovation and improvement in the treatment of childhood cancer: Lessons from high-income countries. Lancet Oncol. 2013, 14, e95–e103. [Google Scholar] [CrossRef]
- Hunger, S.P. Expanding clinical trial networks in pediatric acute lymphoblastic leukemia. J. Clin. Oncol. 2014, 32, 169–170. [Google Scholar] [CrossRef]
- Tsuchida, M.; Ohara, A.; Manabe, A.; Kumagai, M.; Shimada, H.; Kikuchi, A.; Mori, T.; Saito, M.; Akiyama, M.; Fukushima, T.; et al. Long-term results of tokyo children’s cancer study group trials for childhood acute lymphoblastic leukemia, 1984–1999. Leukemia 2010, 24, 383–396. [Google Scholar] [CrossRef]
- Magrath, I.; Steliarova-Foucher, E.; Epelman, S.; Ribeiro, R.C.; Harif, M.; Li, C.K.; Kebudi, R.; Macfarlane, S.D.; Howard, S.C. Paediatric cancer in low-income and middle-income countries. Lancet Oncol. 2013, 14, e104–e116. [Google Scholar] [CrossRef]
- Moricke, A.; Reiter, A.; Zimmermann, M.; Gadner, H.; Stanulla, M.; Dordelmann, M.; Loning, L.; Beier, R.; Ludwig, W.D.; Ratei, R.; et al. Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: Treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial all-bfm 95. Blood 2008, 111, 4477–4489. [Google Scholar] [CrossRef]
- Flohr, T.; Schrauder, A.; Cazzaniga, G.; Panzer-Grumayer, R.; van der Velden, V.; Fischer, S.; Stanulla, M.; Basso, G.; Niggli, F.K.; Schafer, B.W.; et al. Minimal residual disease-directed risk stratification using real-time quantitative PCR analysis of immunoglobulin and T-cell receptor gene rearrangements in the international multicenter trial AIEOP-BFM ALL 2000 for childhood acute lymphoblastic leukemia. Leukemia 2008, 22, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Schrappe, M.; Valsecchi, M.G.; Bartram, C.R.; Schrauder, A.; Panzer-Grumayer, R.; Moricke, A.; Parasole, R.; Zimmermann, M.; Dworzak, M.; Buldini, B.; et al. Late mrd response determines relapse risk overall and in subsets of childhood T-cell all: Results of the AIEOP-BFM-ALL 2000 study. Blood 2011, 118, 2077–2084. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.V.; Ensor, H.M.; Richards, S.M.; Chilton, L.; Schwab, C.; Kinsey, S.E.; Vora, A.; Mitchell, C.D.; Harrison, C.J. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: Results from the UK medical research council ALL97/99 randomised trial. Lancet Oncol. 2010, 11, 429–438. [Google Scholar] [CrossRef]
- Gupta, S.K.; Bakhshi, S.; Chopra, A.; Kamal, V.K. Molecular genetic profile in BCR-ABL1 negative pediatric B-cell acute lymphoblastic leukemia can further refine outcome prediction in addition to that by end-induction minimal residual disease detection. Leuk. Lymphoma 2018, 59, 1899–1904. [Google Scholar] [CrossRef] [PubMed]
- Zerkalenkova, E.; Panfyorova, A.; Kazakova, A.; Baryshev, P.; Shelihova, L.; Kalinina, I.; Novichkova, G.; Maschan, M.; Maschan, A.; Olshanskaya, Y. Molecular characteristic of acute leukemias with t(16;21)/FUS-ERG. Ann. Hematol. 2018, 97, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Vijayakrishnan, J.; Studd, J.; Broderick, P.; Kinnersley, B.; Holroyd, A.; Law, P.J.; Kumar, R.; Allan, J.M.; Harrison, C.J.; Moorman, A.V.; et al. Genome-wide association study identifies susceptibility loci for B-cell childhood acute lymphoblastic leukemia. Nat. Commun. 2018, 9, 1340. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Zhao, X.; Qin, Y.; Zhou, S.; Chang, Y.; Wang, Y.; Zhang, X.; Xu, L.; Huang, X. The prognostic role of E2A-PBX1 expression detected by real-time quantitative reverse transcriptase polymerase chain reaction (RQ-PCR) in b cell acute lymphoblastic leukemia after allogeneic hematopoietic stem cell transplantation. Ann. Hematol. 2018, 97, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic diagnosis of mendelian disorders via RNA sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef]
- Qian, Z.; Liu, H.; Li, M.; Shi, J.; Li, N.; Zhang, Y.; Zhang, X.; Lv, J.; Xie, X.; Bai, Y.; et al. Potential diagnostic power of blood circular RNA expression in active pulmonary tuberculosis. EBioMedicine 2018, 27, 18–26. [Google Scholar] [CrossRef]
- Chen, L.; Lu, D.; Sun, K.; Xu, Y.; Hu, P.; Li, X.; Xu, F. Identification of biomarkers associated with diagnosis and prognosis of colorectal cancer patients based on integrated bioinformatics analysis. Gene 2019, 692, 119–125. [Google Scholar] [CrossRef]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Sci. Rep. 2017, 7, 2409. [Google Scholar] [CrossRef] [PubMed]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.Y.; Yu, Y.; Xie, M.; Yang, M.H.; Zhu, S.; Yang, L.C.; Kang, R.; Tang, D.L.; Zhao, L.L.; Cao, L.Z. Digital gene expression profiling analysis of childhood acute lymphoblastic leukemia. Mol. Med. Rep. 2016, 13, 4321–4328. [Google Scholar] [CrossRef] [PubMed]
- Sakhinia, E.; Estiar, M.A.; Andalib, S.; Rezamand, A. Expression profiling of microarray gene signatures in acute and chronic myeloid leukaemia in human bone marrow. Iran. J. Pediatr. Hematol. Oncol. 2015, 5, 27–42. [Google Scholar]
- Zhao, G.; Jiang, T.; Liu, Y.; Huai, G.; Lan, C.; Li, G.; Jia, G.; Wang, K.; Yang, M. Droplet digital PCR-based circulating microRNA detection serve as a promising diagnostic method for gastric cancer. BMC Cancer 2018, 18, 676. [Google Scholar] [CrossRef] [PubMed]
- Villegas-Ruiz, V.; Moreno, J.; Jacome-Lopez, K.; Zentella-Dehesa, A.; Juarez-Mendez, S. Quality control usage in high-density microarrays reveals differential gene expression profiles in ovarian cancer. Asian Pac. J. Cancer Prev. 2016, 17, 2519–2525. [Google Scholar] [PubMed]
- Villegas-Ruiz, V.; Juarez-Mendez, S. Data mining for identification of molecular targets in ovarian cancer. Asian Pac. J. Cancer Prev. 2016, 17, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Rodriguez, R.A.; Davila-Borja, V.M.; Juarez-Mendez, S. Data mining of pediatric medulloblastoma microarray expression reveals a novel potential subdivision of the group 4 molecular subgroup. Oncol. Lett. 2018, 15, 6241–6250. [Google Scholar]
- Wee, Y.; Liu, Y.; Bhyan, S.B.; Lu, J.; Zhao, M. The pan-cancer analysis of gain-of-functional mutations to identify the common oncogenic signatures in multiple cancers. Gene 2019, 697, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; The Cancer Genome Atlas Research Network; et al. Comprehensive analysis of alternative splicing across tumors from 8705 patients. Cancer Cell 2018, 34, 211–224. [Google Scholar] [CrossRef]
- Bustin, S.A.; Beaulieu, J.F.; Huggett, J.; Jaggi, R.; Kibenge, F.S.; Olsvik, P.A.; Penning, L.C.; Toegel, S. Miqe precis: Practical implementation of minimum standard guidelines for fluorescence-based quantitative real-time PCR experiments. BMC Mol. Biol. 2010, 11, 74. [Google Scholar] [CrossRef]
- Bustin, S.A. Quantification of mrna using real-time reverse transcription PCR (RT-PCR): Trends and problems. J. Mol. Endocrinol. 2002, 29, 23–39. [Google Scholar] [CrossRef]
- Bustin, S.A.; Nolan, T. Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J. Biomol. Tech. 2004, 15, 155–166. [Google Scholar]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The digital MIQE guidelines: Minimum information for publication of quantitative digital PCR experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef]
- Sanders, R.; Huggett, J.F.; Bushell, C.A.; Cowen, S.; Scott, D.J.; Foy, C.A. Evaluation of digital PCR for absolute DNA quantification. Anal. Chem. 2011, 83, 6474–6484. [Google Scholar] [CrossRef]
- Sanders, R.; Mason, D.J.; Foy, C.A.; Huggett, J.F. Evaluation of digital PCR for absolute RNA quantification. PLoS ONE 2013, 8, e75296. [Google Scholar] [CrossRef]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef]
- Dube, S.; Qin, J.; Ramakrishnan, R. Mathematical analysis of copy number variation in a DNA sample using digital PCR on a nanofluidic device. PLoS ONE 2008, 3, e2876. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin. Cancer Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef]
- Mencia-Trinchant, N.; Hu, Y.; Alas, M.A.; Ali, F.; Wouters, B.J.; Lee, S.; Ritchie, E.K.; Desai, P.; Guzman, M.L.; Roboz, G.J.; et al. Minimal residual disease monitoring of acute myeloid leukemia by massively multiplex digital PCR in patients with npm1 mutations. J. Mol. Diagn. 2017, 19, 537–548. [Google Scholar] [CrossRef]
- Stahlberg, A.; Kubista, M.; Pfaffl, M. Comparison of reverse transcriptases in gene expression analysis. Clin. Chem. 2004, 50, 1678–1680. [Google Scholar] [CrossRef] [PubMed]
- Demeke, T.; Eng, M. Effect of endogenous reference genes on digital PCR assessment of genetically engineered canola events. Biomol. Detect. Quantif. 2018, 15, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Valente, V.; Teixeira, S.A.; Neder, L.; Okamoto, O.K.; Oba-Shinjo, S.M.; Marie, S.K.; Scrideli, C.A.; Paco-Larson, M.L.; Carlotti, C.G., Jr. Selection of suitable housekeeping genes for expression analysis in glioblastoma using quantitative RT-PCR. Ann. Neurosci. 2014, 21, 62–63. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequences 5′ → 3′ | Amplicon Size (bp) | Tm (°C) |
|---|---|---|---|
| ACTB | Fw 5′-TCACAATGTGGCCGAGGACTTT-3′ Rv 5′-AGAAGTGGGGTGGCTTTTAGGATG-3′ | 115 | 60 |
| GAPDH | Fw 5′-CTCAACGACCACTTTGTCAAGCTC-3′ Rv 5′-CTCTCTTCCTCTTGTGCTCTTGCT-3′ | 147 | 60 |
| RPL4-a | Fw 5′-CGAATGAGAGCTGGCAAAGGCAAA-3′ Rv 5′-ACGCCAAGTGCCGTACAATTCATC-3′ | 243 | 60 |
| RPL4 | Fw 5′- GTGGGACGTTTCTGCATTTG-3′ Rv 5′-TGTGCATGGGAAGATTGTAGT-3′ | 112 | 60 |
| RPS18-a | Fw 5′-AATCCACGCCAGTACAAGATCCCA-3′ Rv 5′-TTTCTTCTTGGACACACCCACGGT-3′ | 241 | 58 |
| RPS18 | Fw 5′- CAGCCAGGTCCTAGCCAATG-3′ Rv 5′-CCATCTATGGGCCCGAATCT-3′ | 82 | 60 |
| UBQLN2 | Fw 5′-CAGCCTGAAGGATCAGTGTAGT-3′ Rv 5′-AGGGTCTCTTTATGGGAGAAGC-3′ | 84 | 60 |
| UQCR2 | Fw 5′-CCTGCGGGGTGATGTTGATA-3′ Rv 5′-CAGCTACTTCCCAACGACGA-3′ | 83 | 60 |
| PGGT1B | Fw 5′-CTGTGGTTTCCGAGGCTCTT-3′ Rv 5′-GCATGAGAGGCCAGTGTAGG-3′ | 118 | 60 |
| PSMB6 | Fw 5′-TGACACCTATTCACGACCGC-3′ Rv 5′-GGACCAGTGGAGGCTCATTC-3′ | 129 | 60 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villegas-Ruíz, V.; Olmos-Valdez, K.; Castro-López, K.A.; Saucedo-Tepanecatl, V.E.; Ramírez-Chiquito, J.C.; Pérez-López, E.I.; Medina-Vera, I.; Juárez-Méndez, S. Identification and Validation of Novel Reference Genes in Acute Lymphoblastic Leukemia for Droplet Digital PCR. Genes 2019, 10, 376. https://doi.org/10.3390/genes10050376
Villegas-Ruíz V, Olmos-Valdez K, Castro-López KA, Saucedo-Tepanecatl VE, Ramírez-Chiquito JC, Pérez-López EI, Medina-Vera I, Juárez-Méndez S. Identification and Validation of Novel Reference Genes in Acute Lymphoblastic Leukemia for Droplet Digital PCR. Genes. 2019; 10(5):376. https://doi.org/10.3390/genes10050376
Chicago/Turabian StyleVillegas-Ruíz, Vanessa, Karina Olmos-Valdez, Kattia Alejandra Castro-López, Victoria Estefanía Saucedo-Tepanecatl, Josselen Carina Ramírez-Chiquito, Eleazar Israel Pérez-López, Isabel Medina-Vera, and Sergio Juárez-Méndez. 2019. "Identification and Validation of Novel Reference Genes in Acute Lymphoblastic Leukemia for Droplet Digital PCR" Genes 10, no. 5: 376. https://doi.org/10.3390/genes10050376
APA StyleVillegas-Ruíz, V., Olmos-Valdez, K., Castro-López, K. A., Saucedo-Tepanecatl, V. E., Ramírez-Chiquito, J. C., Pérez-López, E. I., Medina-Vera, I., & Juárez-Méndez, S. (2019). Identification and Validation of Novel Reference Genes in Acute Lymphoblastic Leukemia for Droplet Digital PCR. Genes, 10(5), 376. https://doi.org/10.3390/genes10050376