Preparation of Nanocomposites of Poly(ε-caprolactone) and Multi-Walled Carbon Nanotubes by Ultrasound Micro-Molding. Influence of Nanotubes on Melting and Crystallization

 ,
,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Nanocomposites
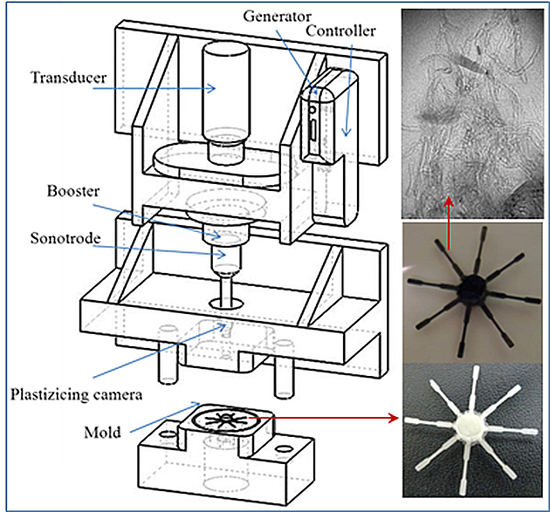
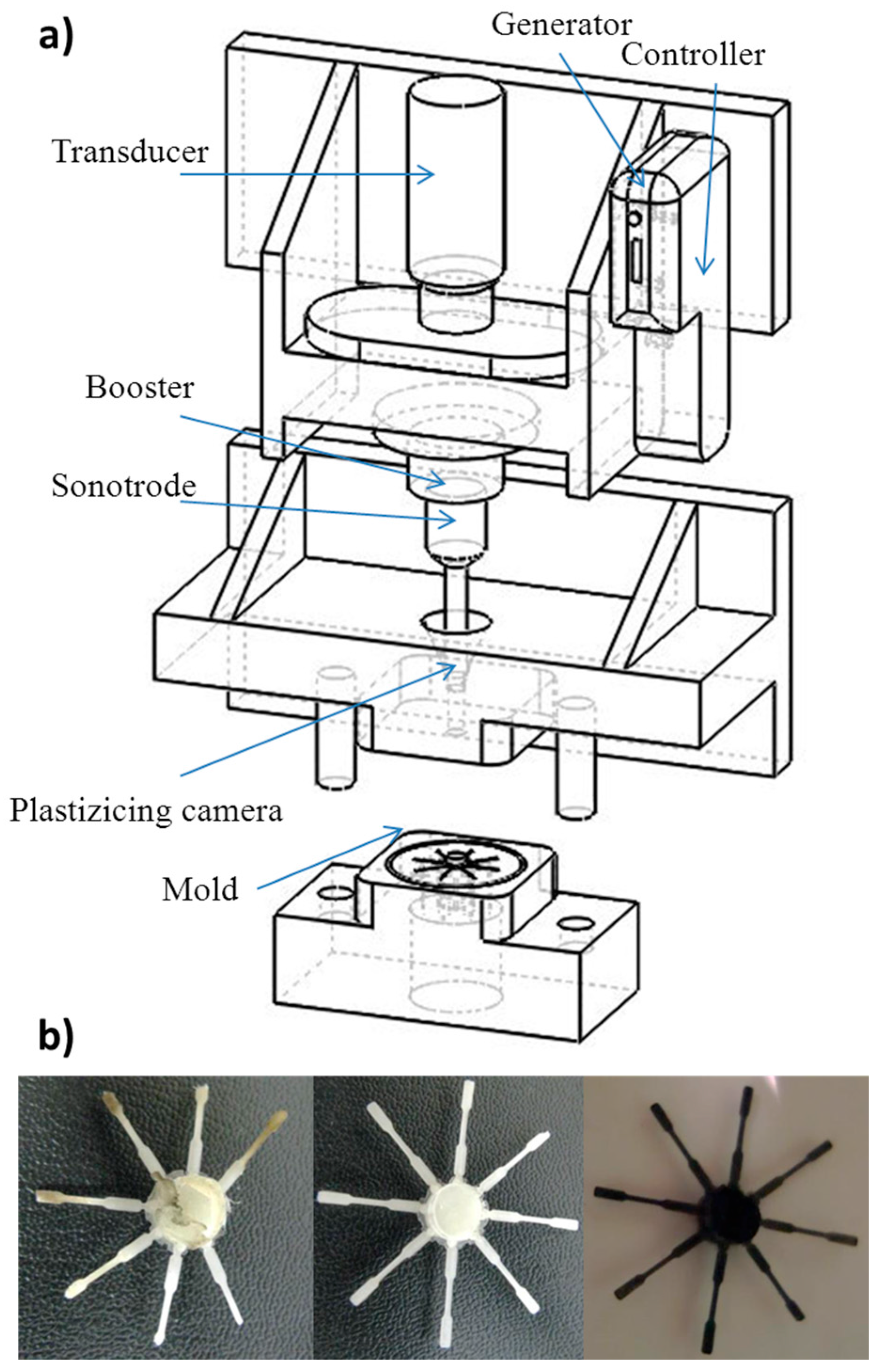
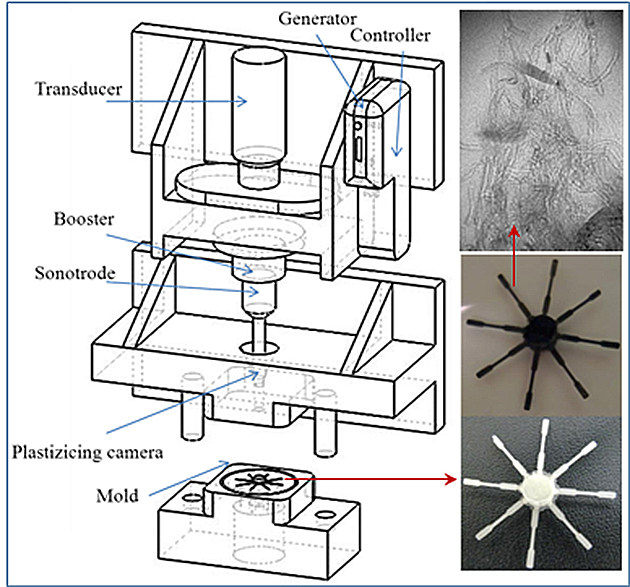
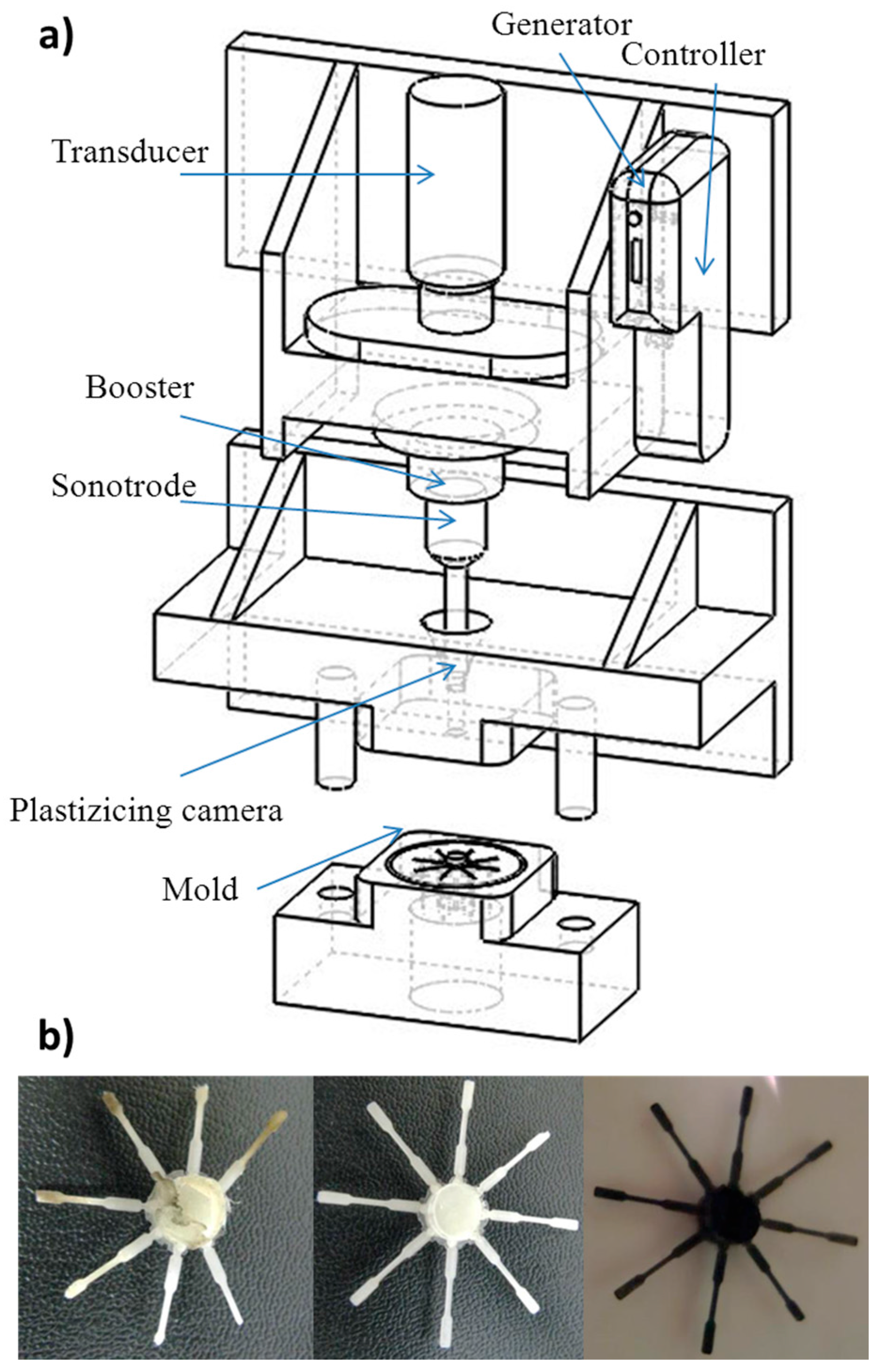
2.3. Micro-Molding Equipment
2.4. Measurements
3. Results and Discussion
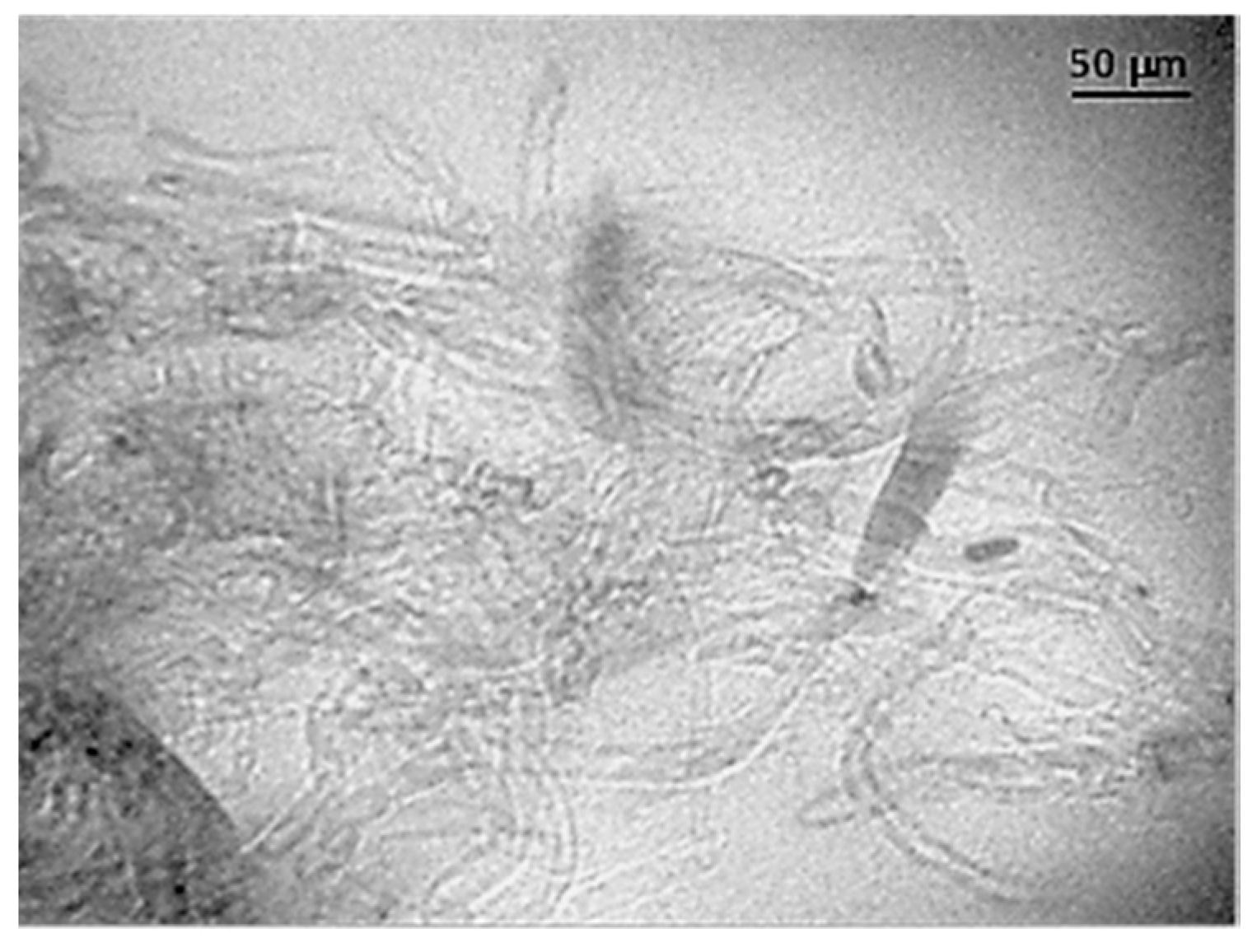
3.1. Ultrasound Micro-Molding of PCL and PCL/MWCNT Nanocomposites
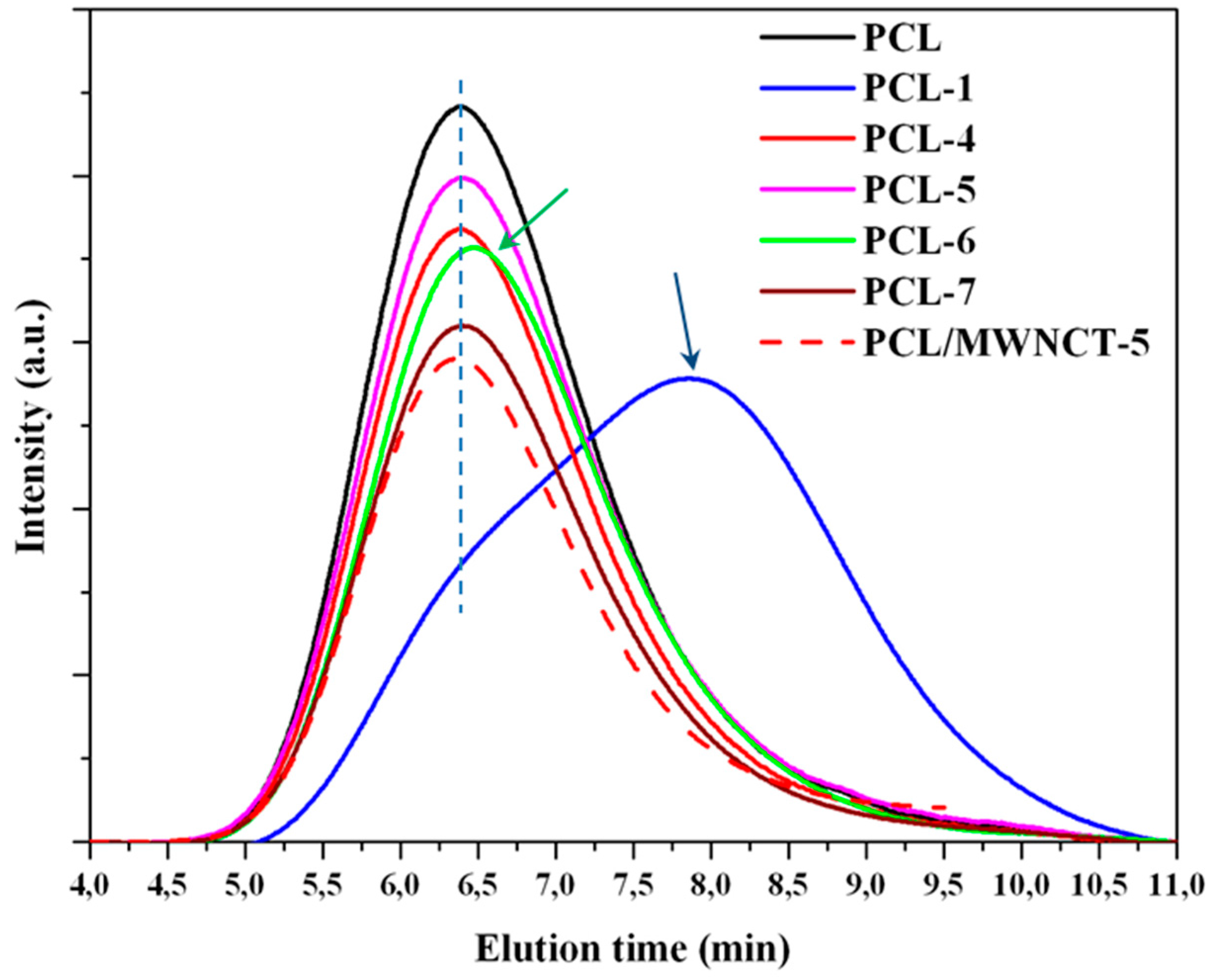
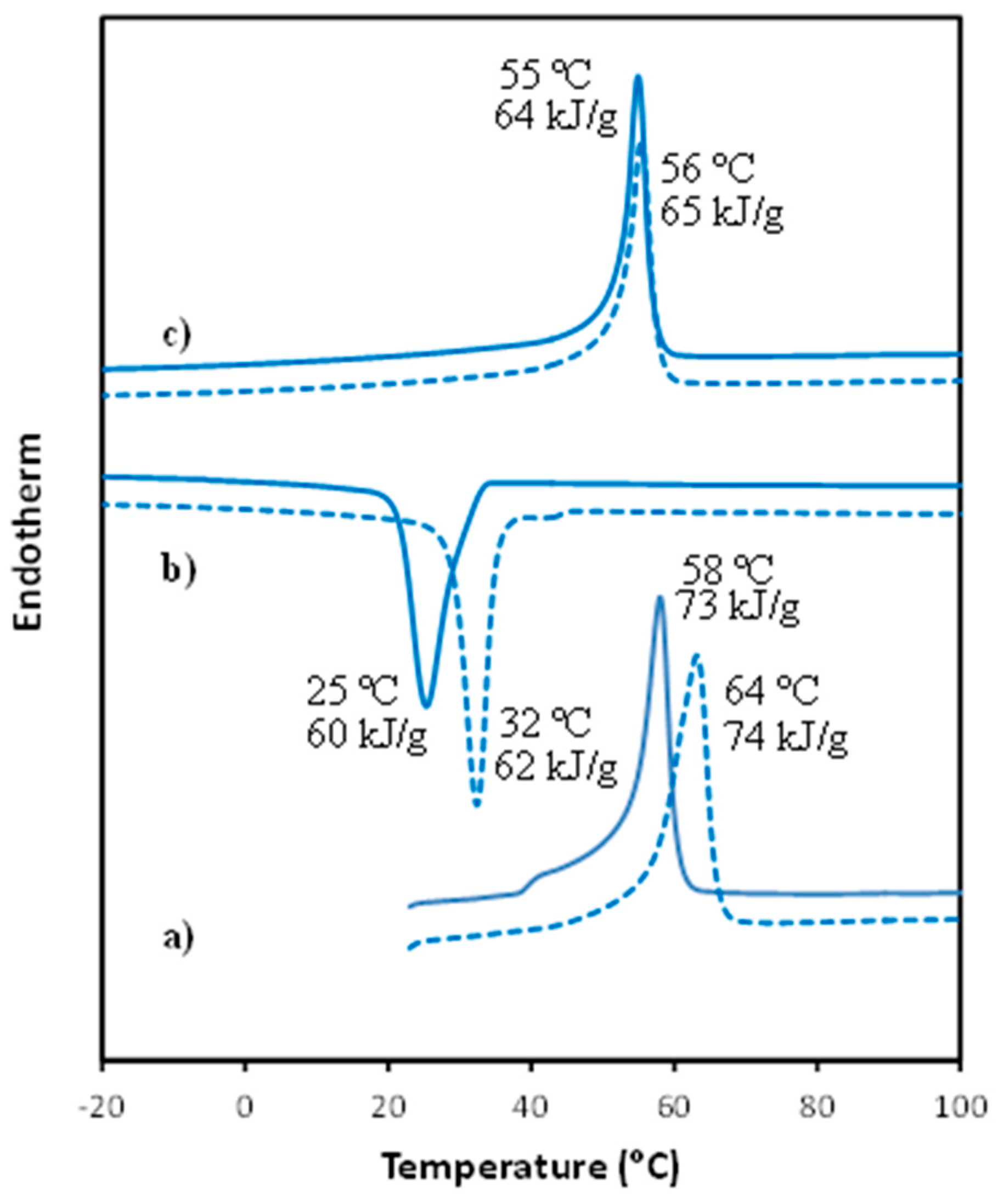
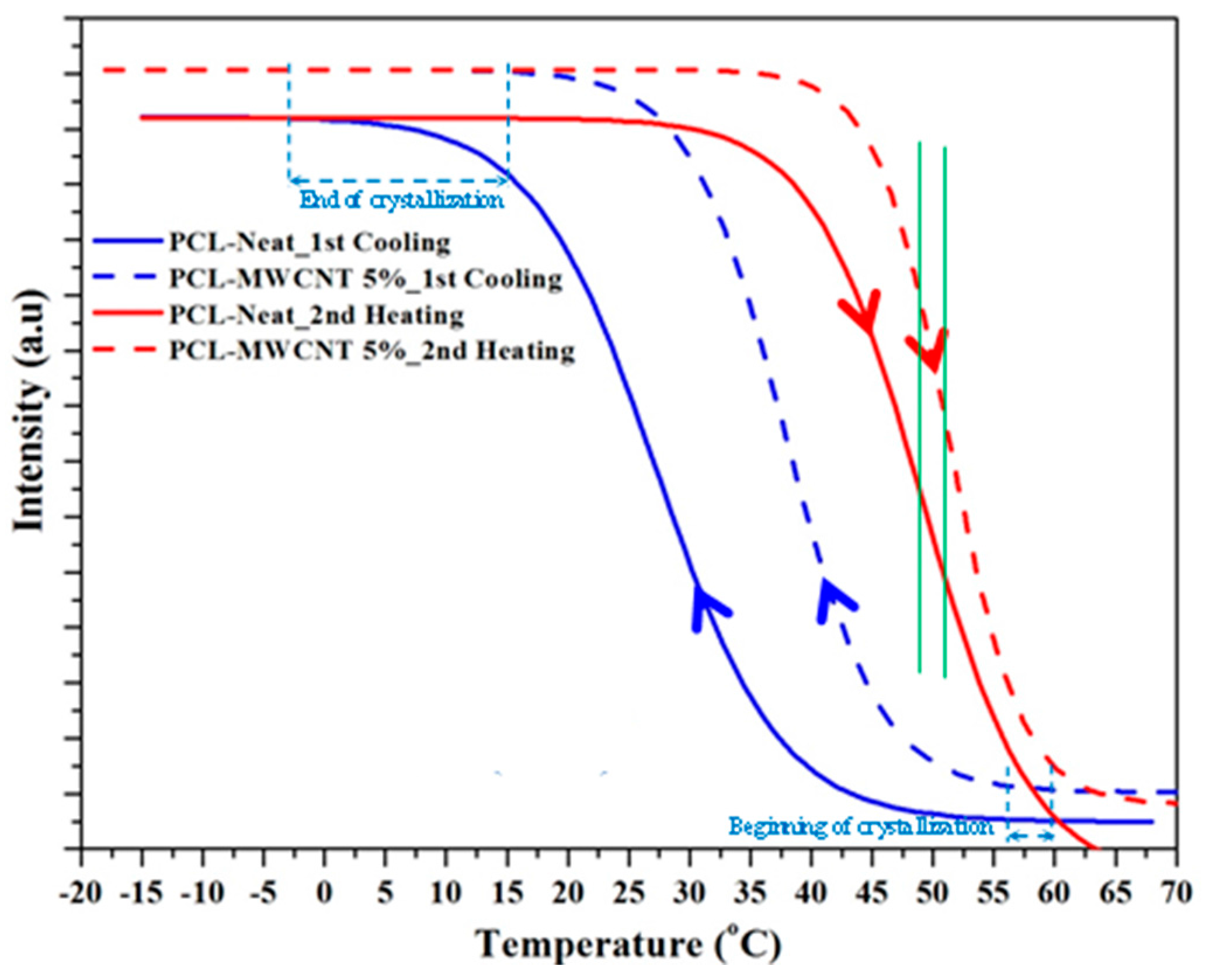
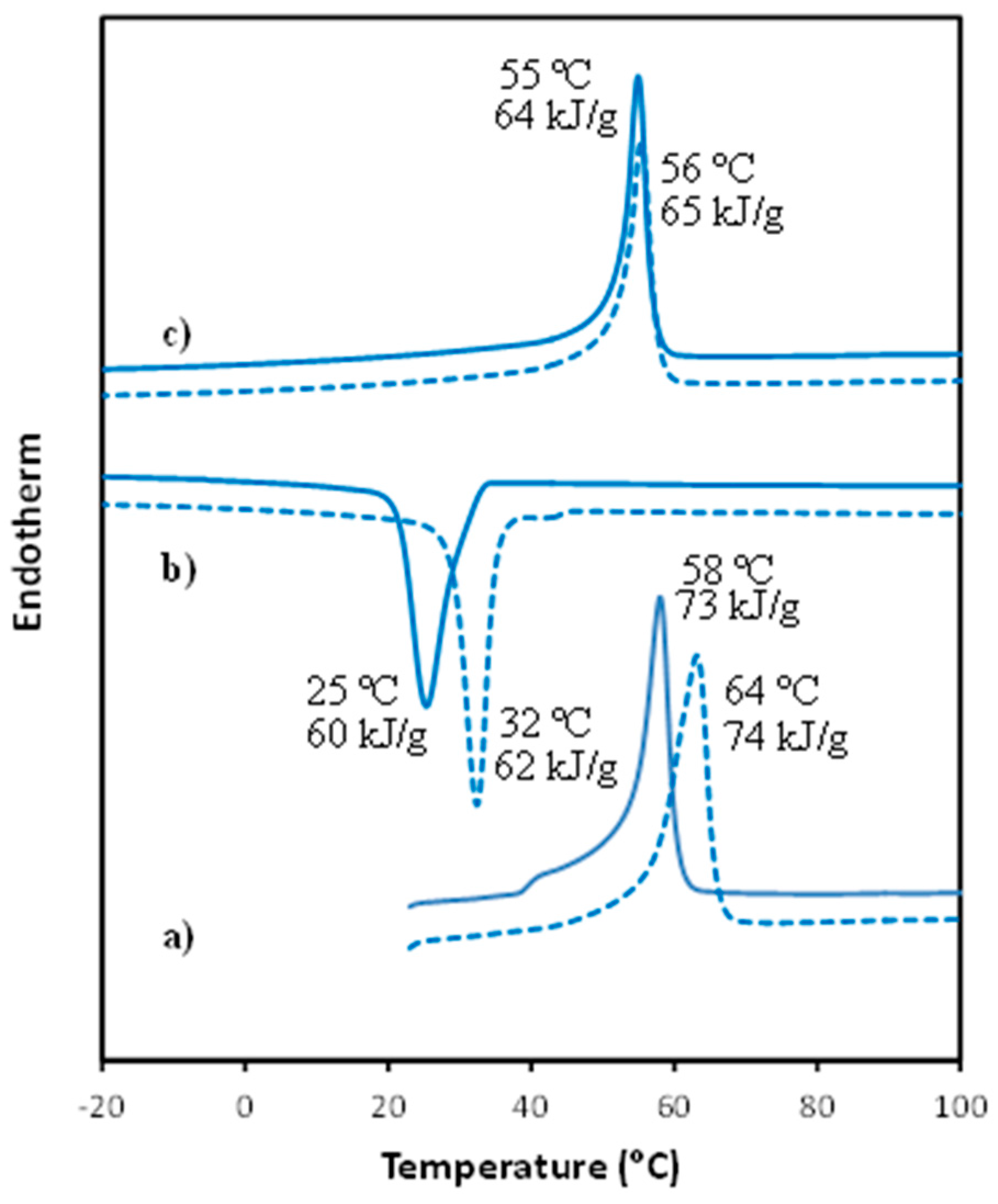
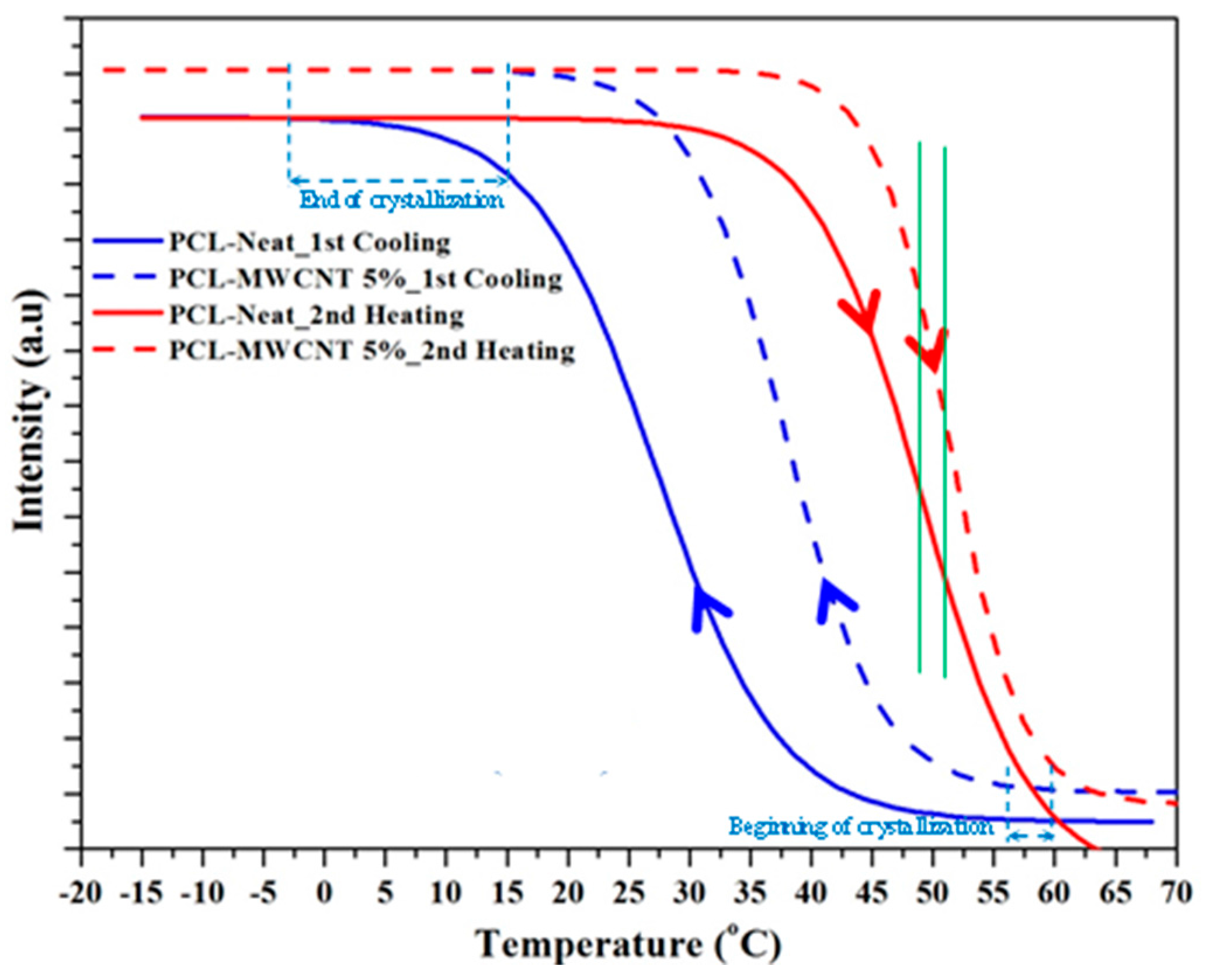
3.2. Thermal Properties of PCL/MWCNT Nanocomposites
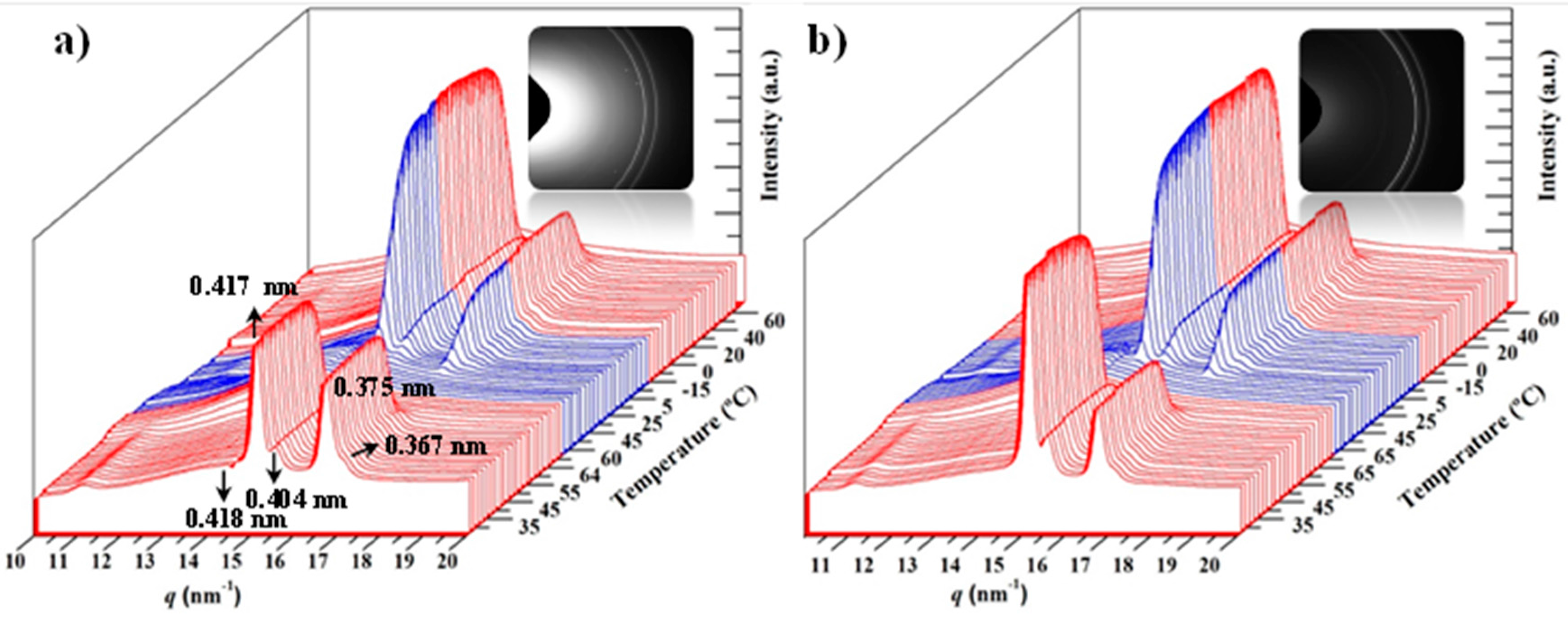
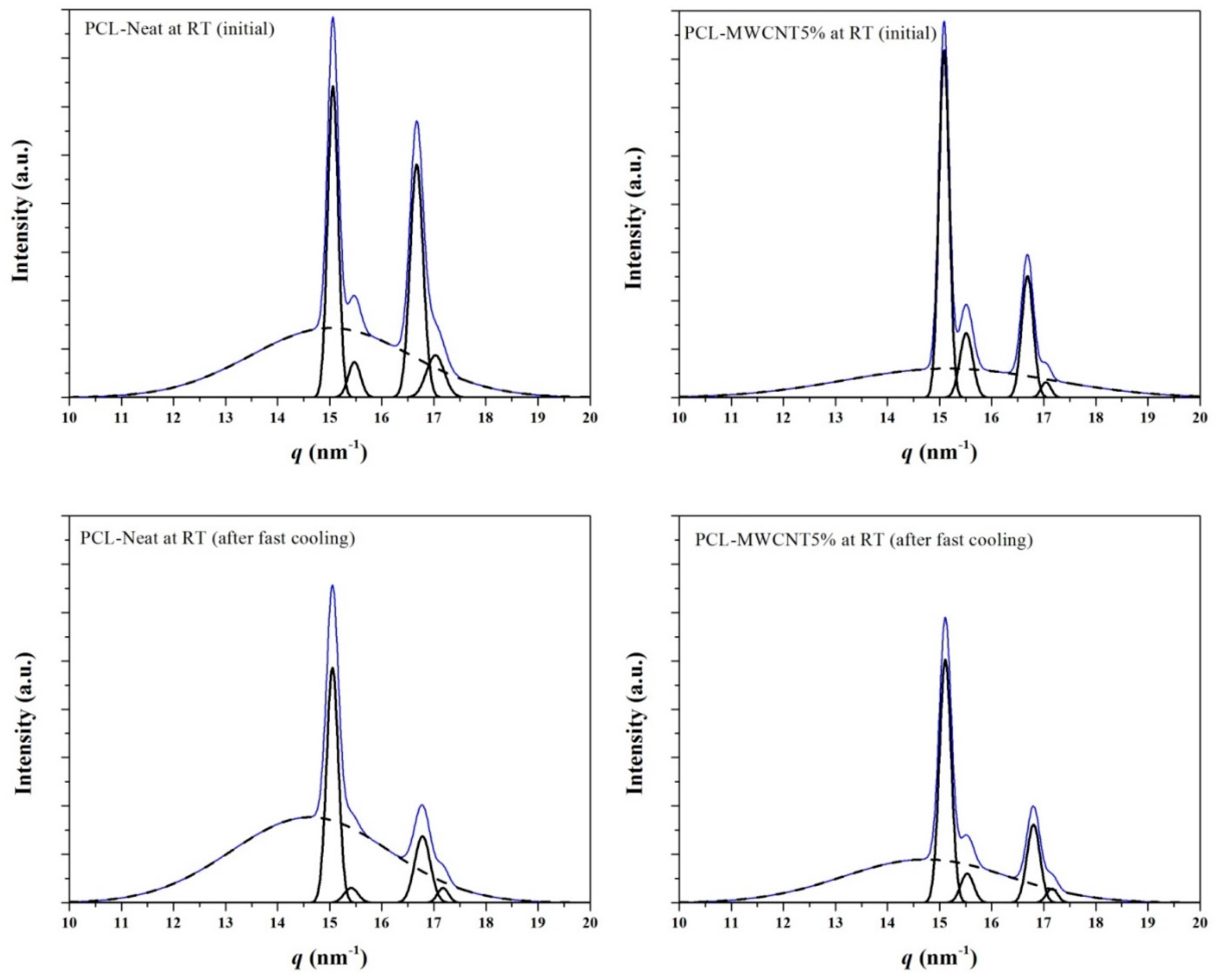
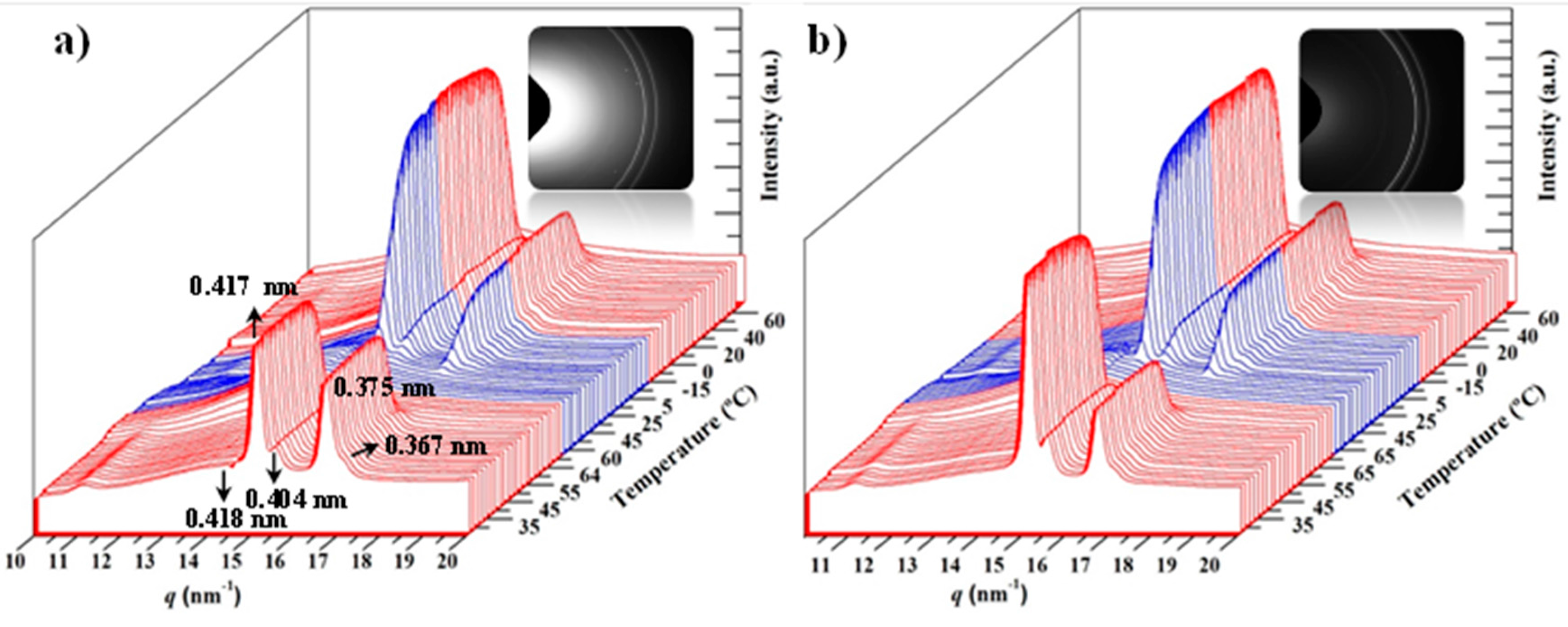
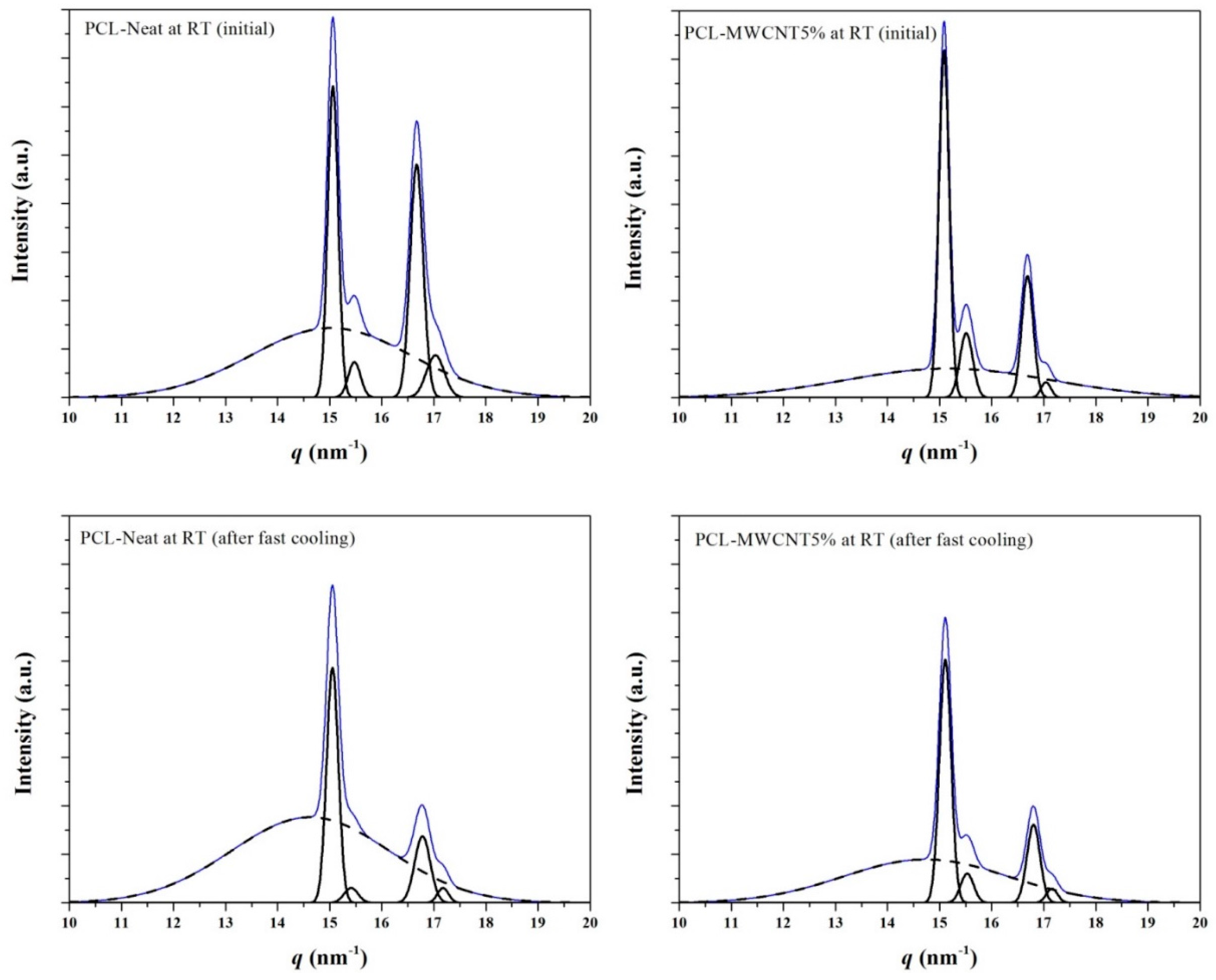
3.3. Melting and Crystallization of PCL and PCL/MWCNT-5 Nanocomposites from Time-Resolved WAXD Experiments
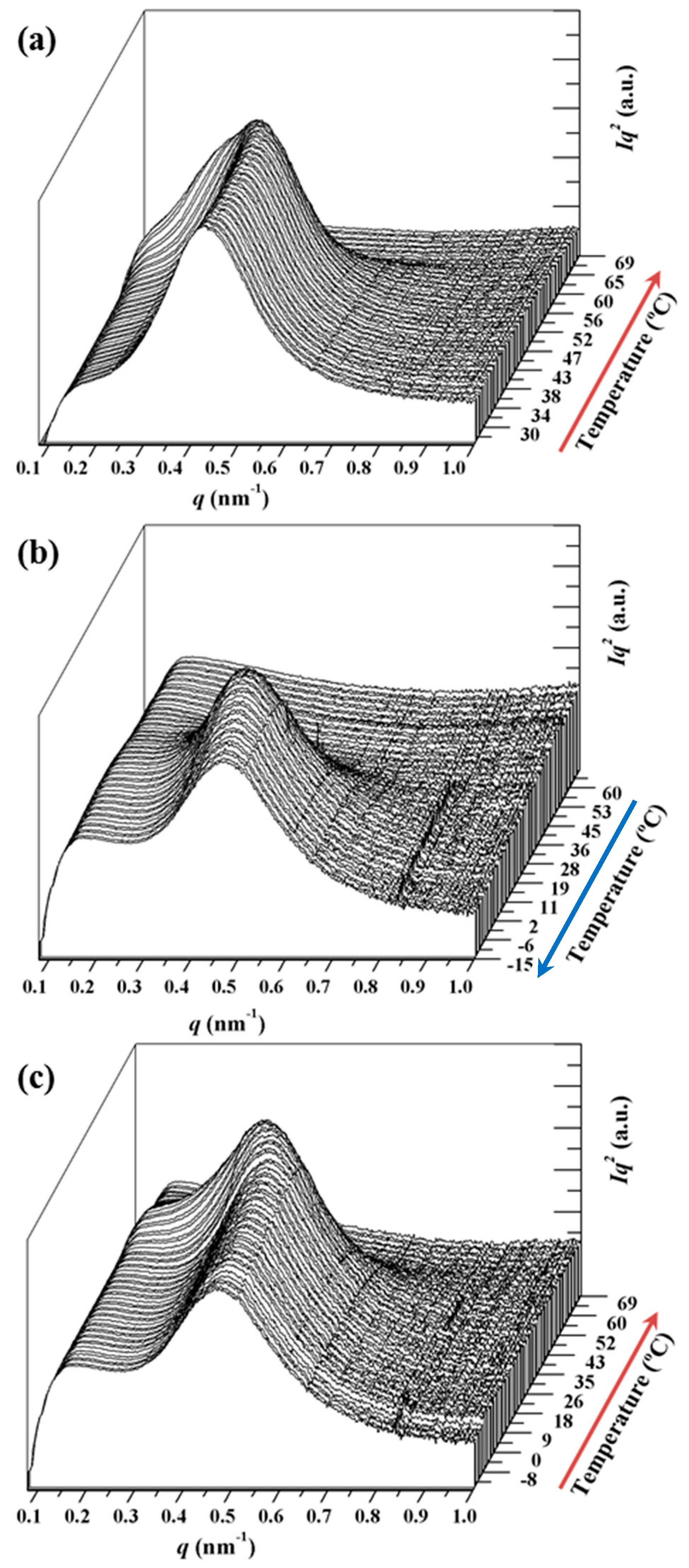
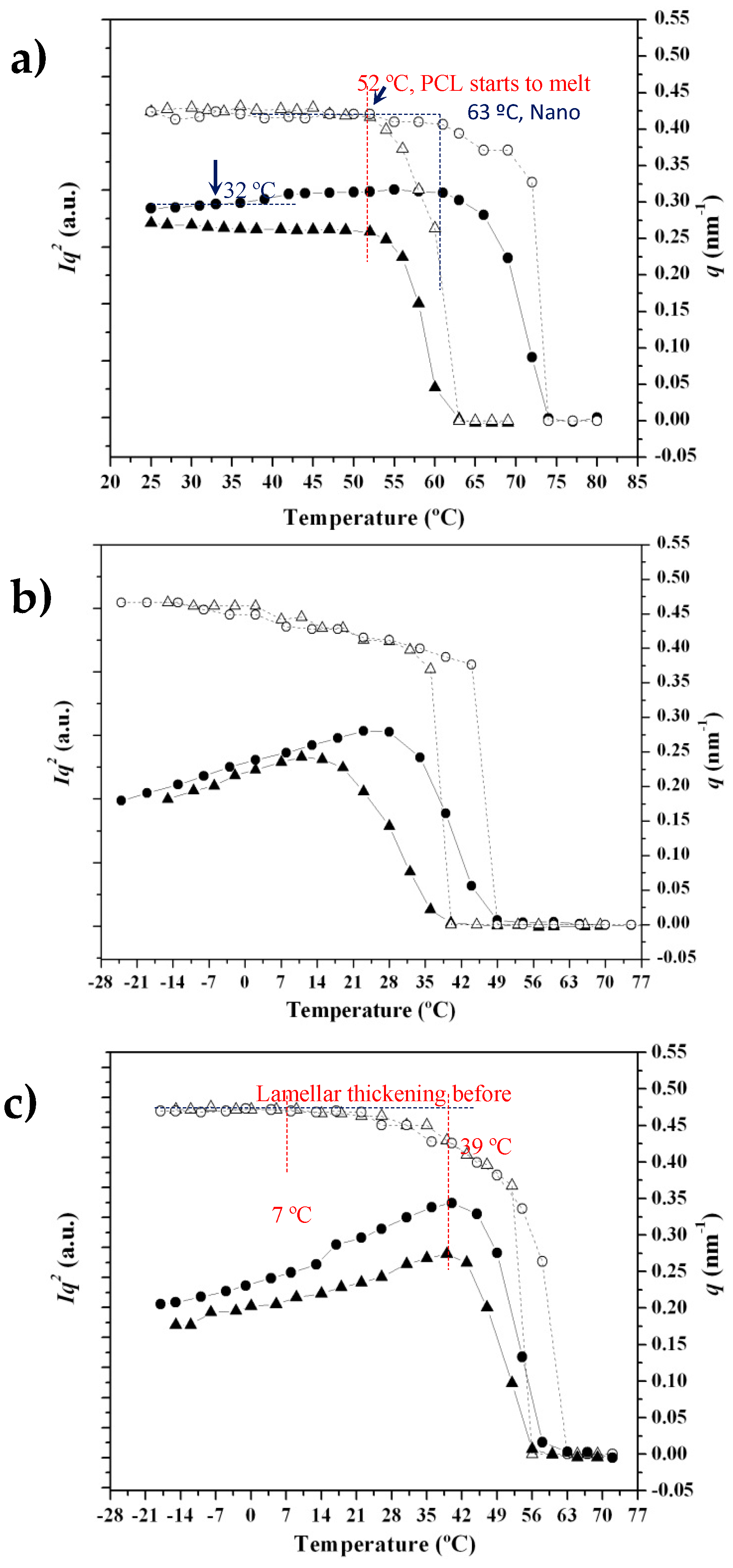
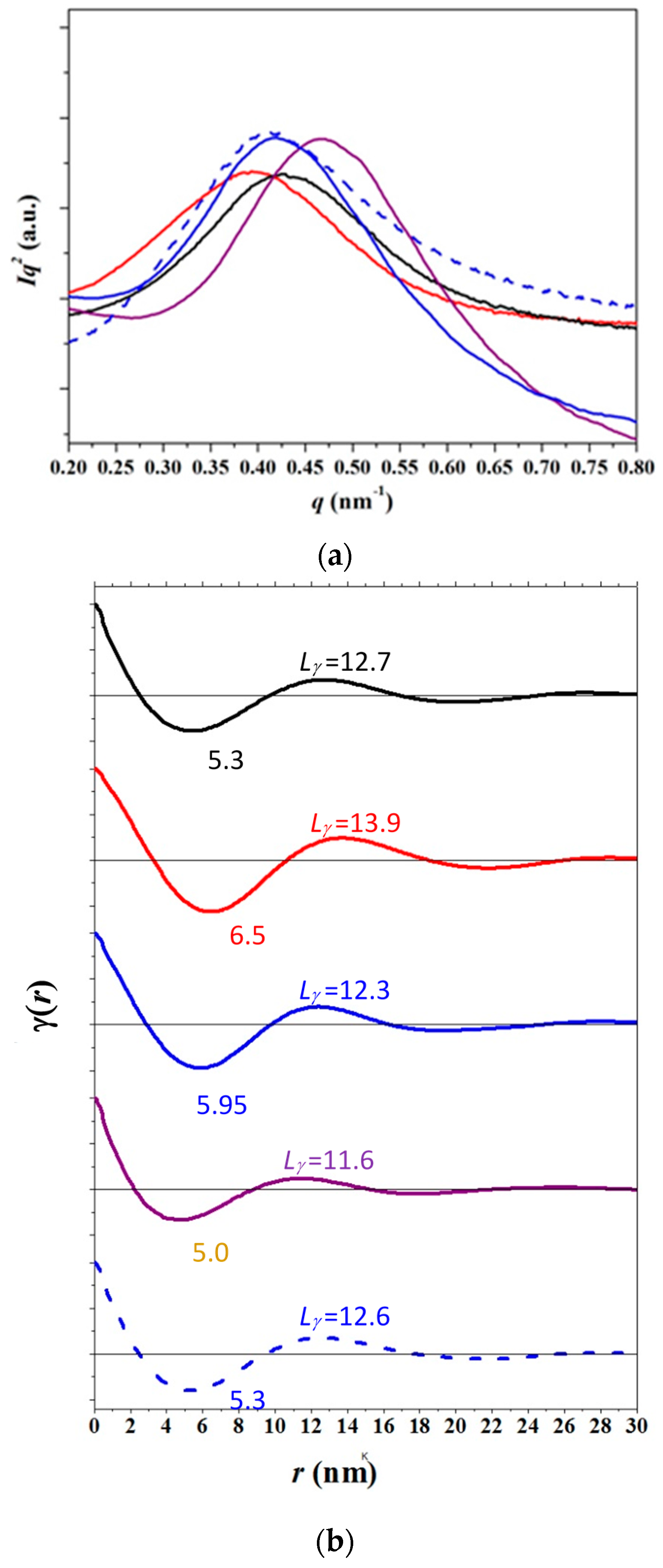
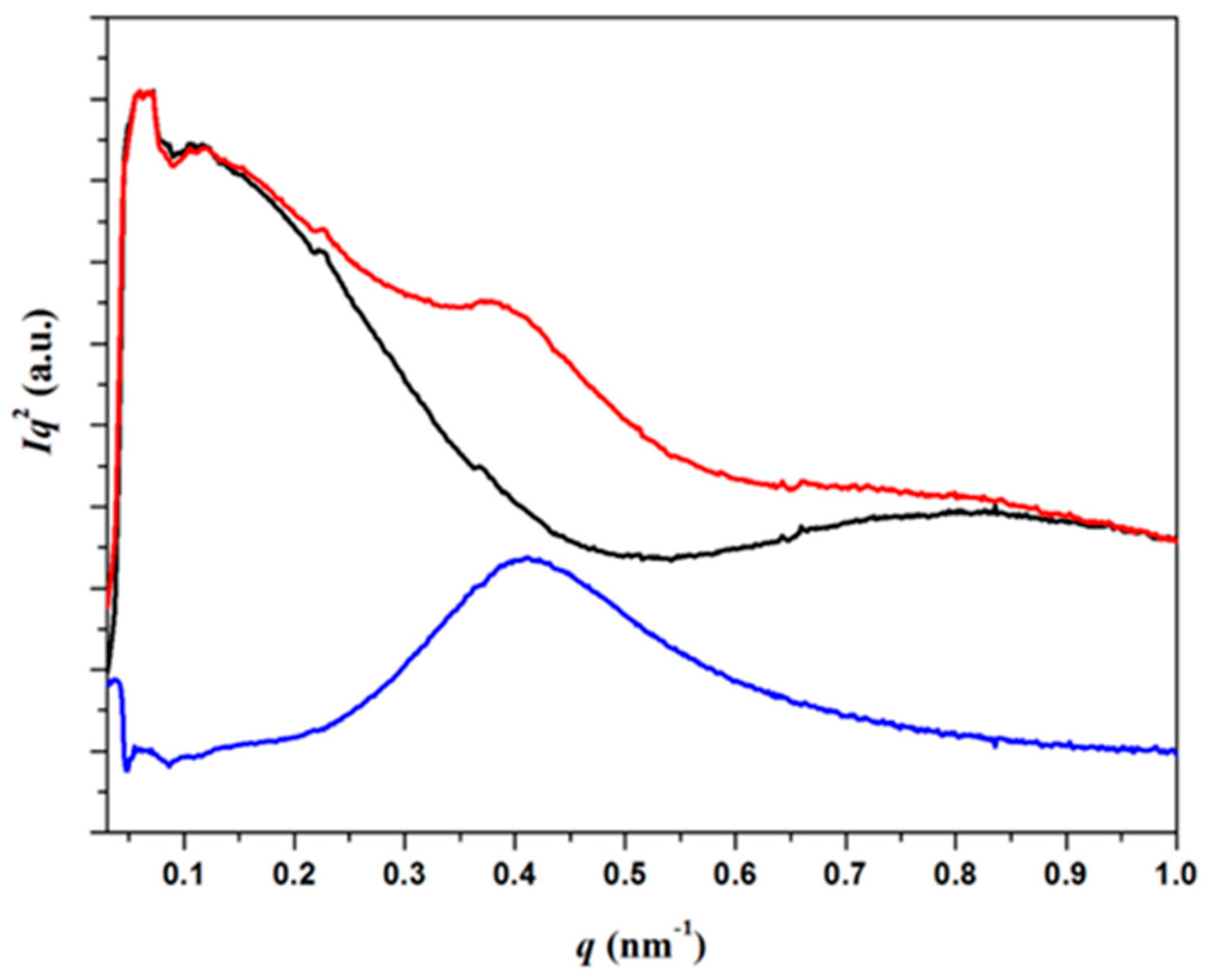
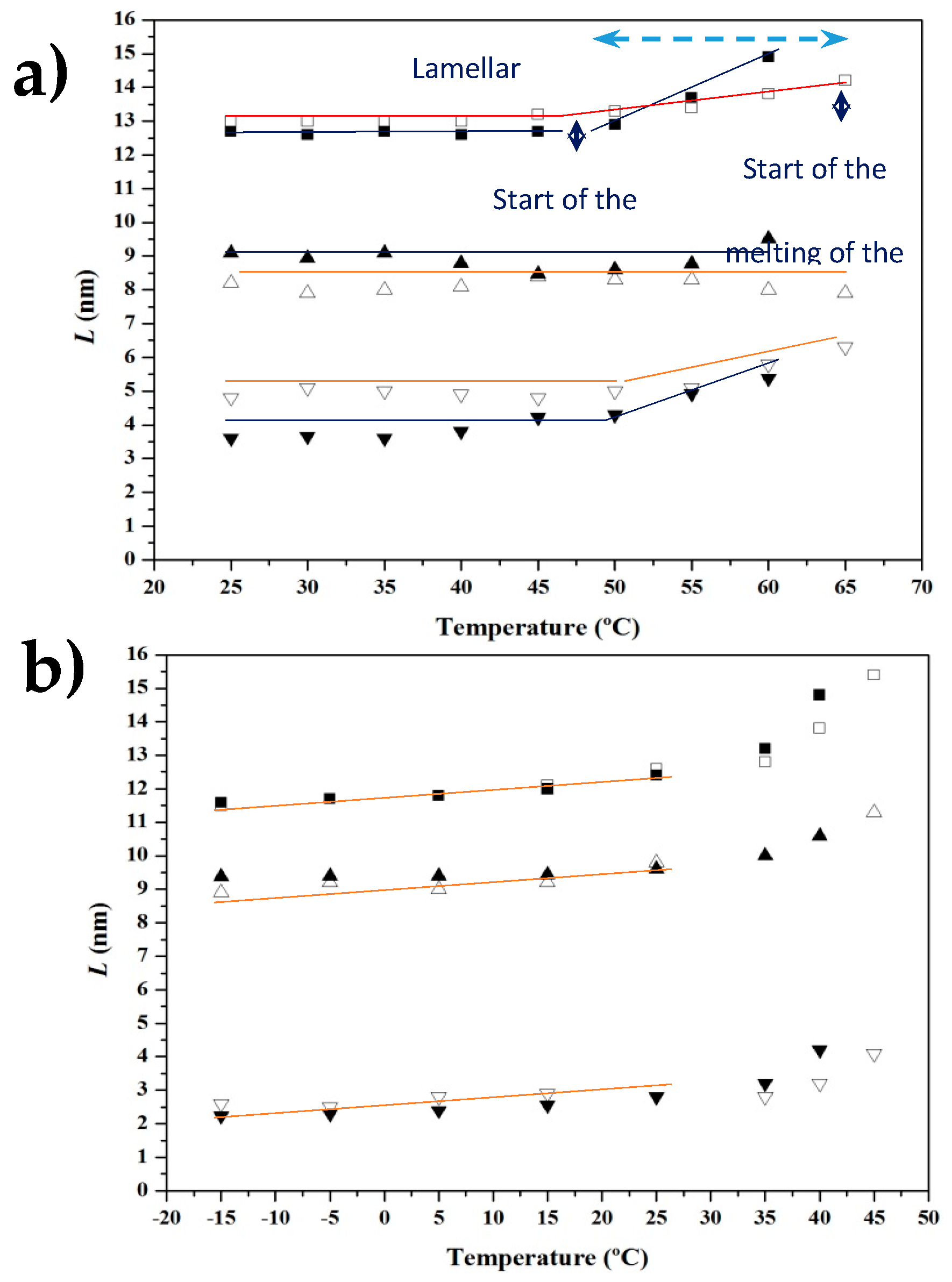
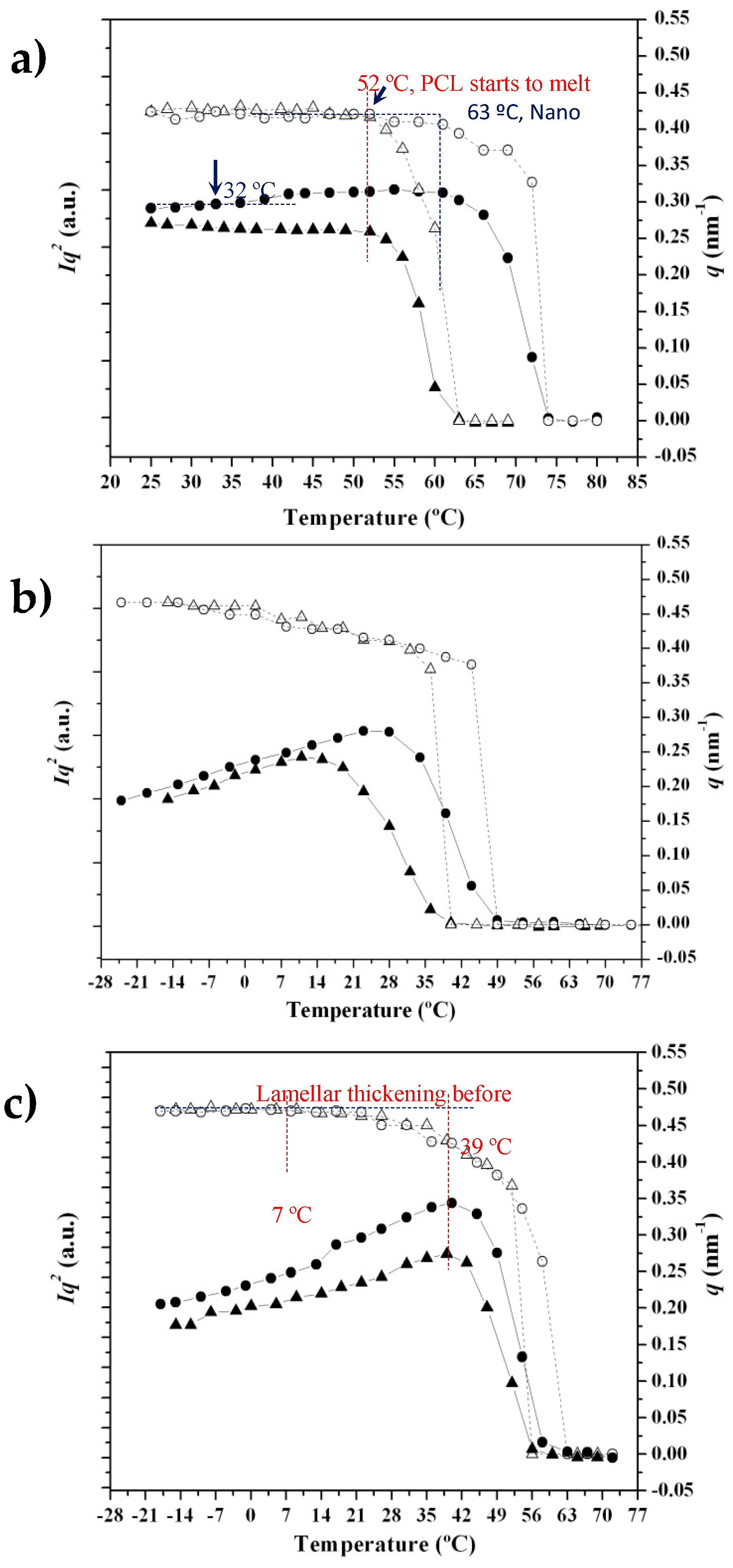
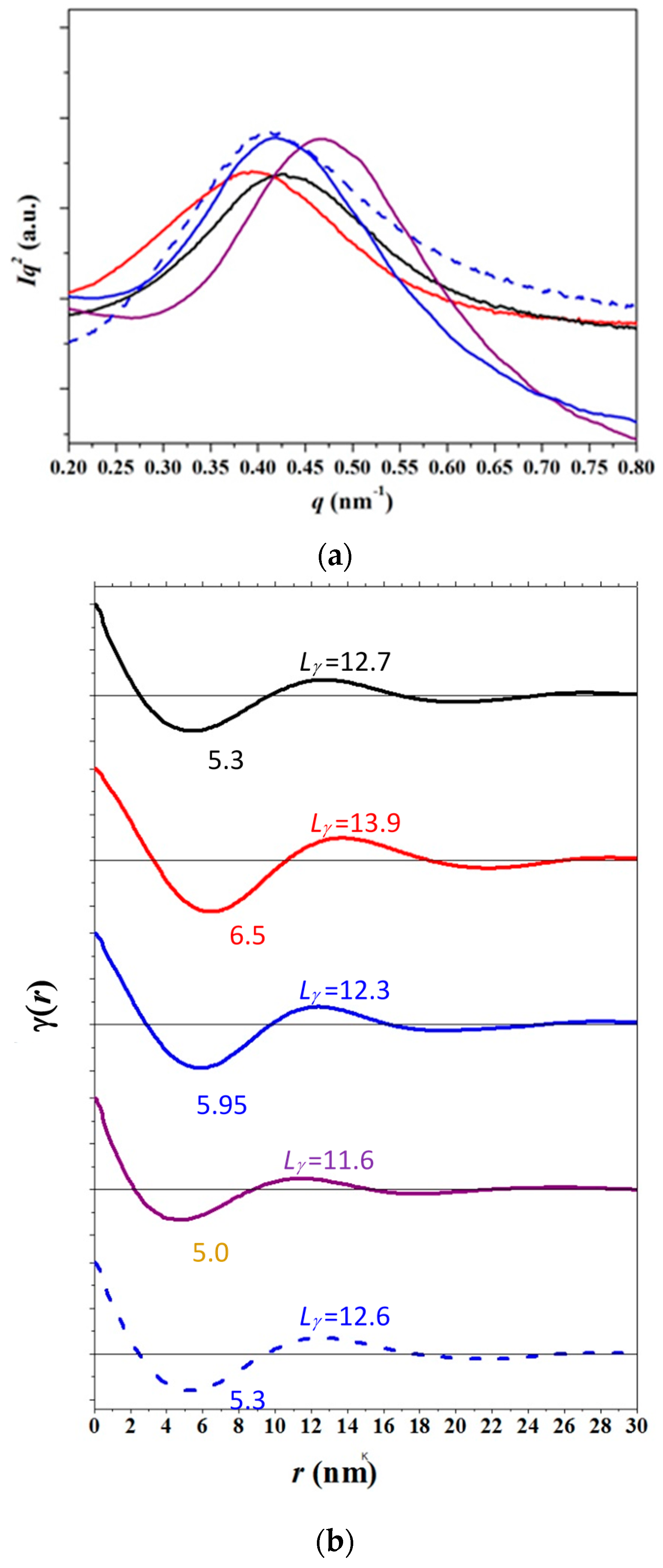
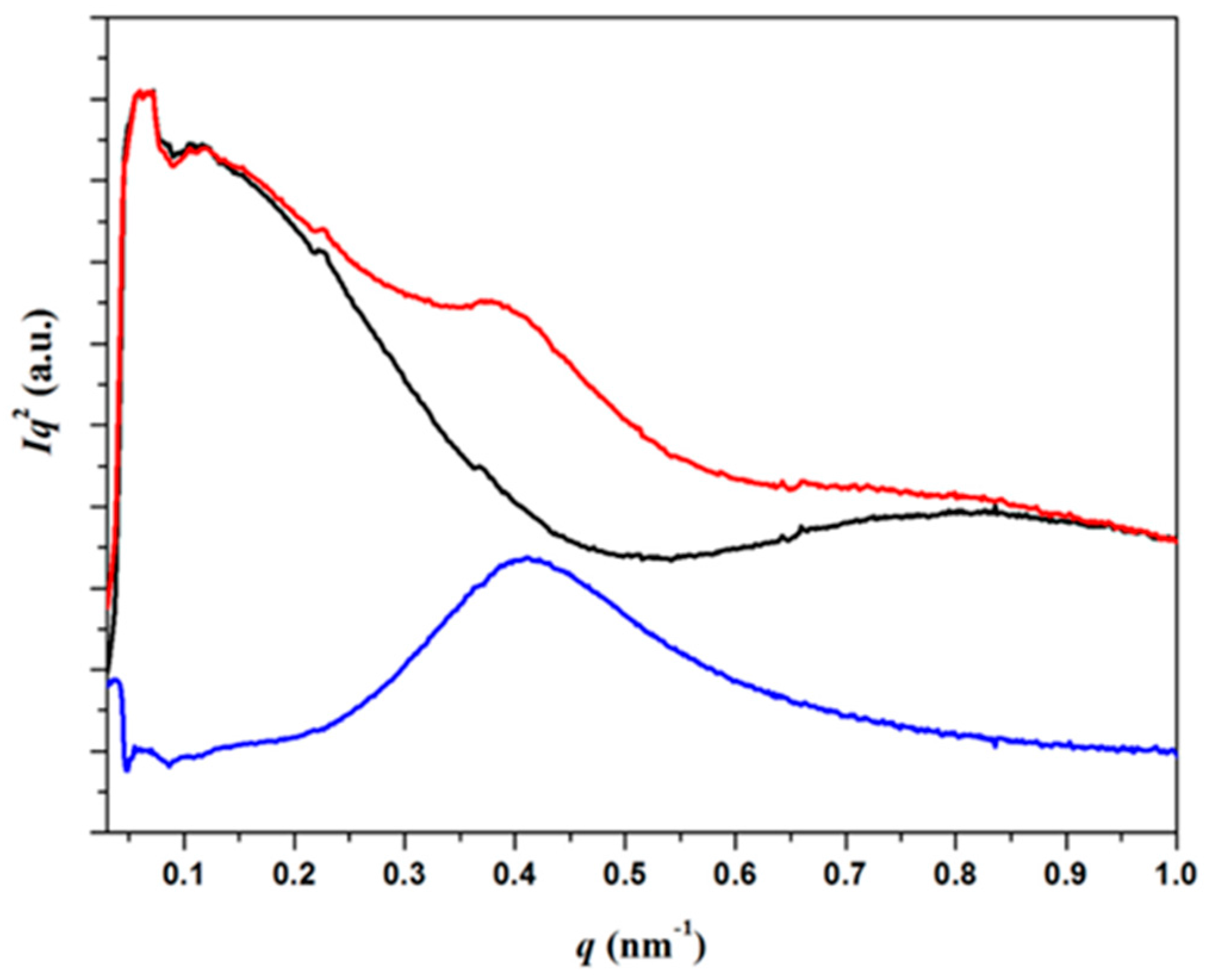
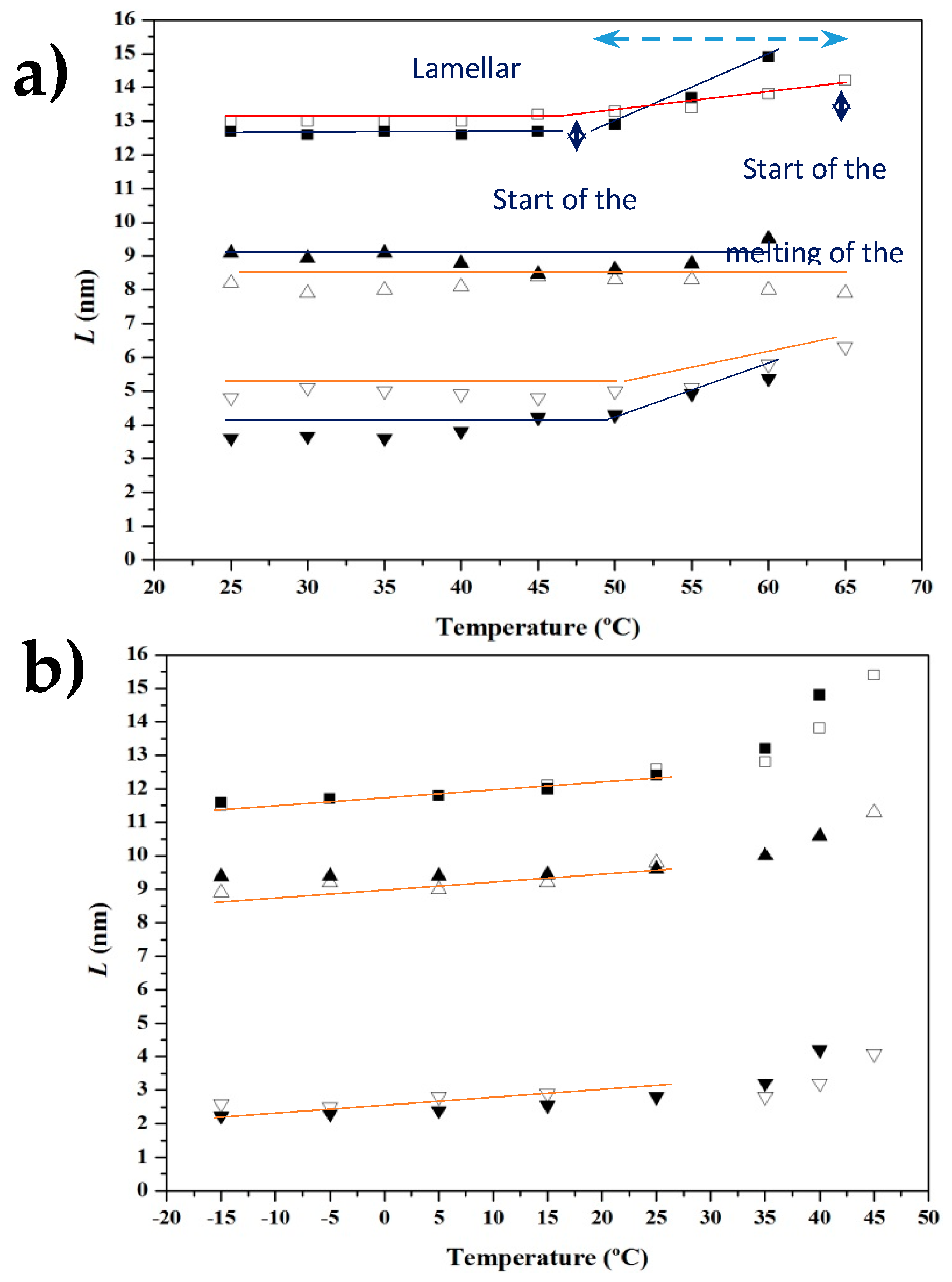
3.4. SAXS Analysis of the Melting Crystallization of PCL and PCL/MWCNT Nanocomposites
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Planellas, M.; Sacristán, M.; Rey, L.; Olmo, C.; Aymamí, J.; Casas, M.T.; del Valle, L.J.; Franco, L.; Puiggalí, J. Micro-molding with ultrasonic vibration energy: New method to disperse nanoclays in polymer matrices. Ultrason. Sonochem. 2014, 21, 1557–1569. [Google Scholar] [CrossRef] [PubMed]
- Díaz, A.; Casas, M.T.; del Valle, L.J.; Aymamí, J.; Olmo, C.; Puiggalí, J. Preparation of micro-molded exfoliated clay nanocomposites by means of ultrasonic technology. J. Polym. Res. 2014, 21, 584–596. [Google Scholar] [CrossRef]
- Díaz, A.; Casas, M.T.; Puiggalí, J. Dispersion of functionalized silica micro- and nanoparticles into poly(nonamethylene azelate) by ultrasonic micro-molding. Appl. Sci. 2015, 5, 1252–1271. [Google Scholar] [CrossRef]
- Michaeli, W.; Spennemann, A.; Gartner, R. New plastification concepts for micro injection moulding. Microsyst. Technol. 2002, 8, 55–57. [Google Scholar] [CrossRef]
- Michaeli, W.; Starke, C. Ultrasonic investigations of the thermoplastics injection moulding process. Polym. Test. 2005, 24, 205–209. [Google Scholar] [CrossRef]
- Michaeli, W.; Kamps, T.; Hopmann, C. Manufacturing of polymer micro parts by ultrasonic plasticization and direct injection. Microsyst. Technol. 2011, 17, 243–249. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Y.; Li, H.; Lai, S.Y.; Jow, J. Physical and chemical effects of ultrasound vibration on polymer melt in extrusion. Ultrason. Sonochem. 2010, 17, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Guo, S.; Li, H. Ultrasonic improvement of rheological behavior of polystyrene. J. Appl. Polym. Sci. 2002, 84, 2451–2460. [Google Scholar] [CrossRef]
- Kang, J.; Chen, J.; Cao, Y.; Li, H. Effects of ultrasound on the conformation and crystallization behavior of isotactic polypropylene and β-isotactic polypropylene. Polymer 2010, 51, 249–256. [Google Scholar] [CrossRef]
- Cao, Y.; Li, H. Influence of ultrasound on the processing and structure of polypropylene during extrusion. Polym. Eng. Sci. 2002, 42, 1534–1540. [Google Scholar] [CrossRef]
- Sacristán, M.; Plantá, X.; Morell, M.; Puiggalí, J. Effects of ultrasonic vibration on the micro-molding processing of polylactide. Ultrason. Sonochem. 2014, 21, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Usuki, A.; Kojima, Y.; Kawasumi, M.; Okada, A.; Fukushima, Y.; Kurauchi, T.; Kamigaito, O. Mechanical properties of nylon 6-clay hybrid. J. Mater. Res. 1993, 8, 1185–1189. [Google Scholar] [CrossRef]
- Kojima, Y.; Usuki, A.; Kawasumi, M.; Okada, A.; Kurauchi, T.; Kamigaito, O. Synthesis of nylon 6-clay hybrid by montmorillonite intercalated with ε-caprolactam. J. Polym. Sci. Part A 1993, 31, 983–986. [Google Scholar] [CrossRef]
- Kojima, Y.; Usuki, A.; Kawasumi, M.; Okada, A.; Kurauchi, T.; Kamigaito, O. One-pot synthesis of nylon 6-clay hybrid. J. Polym. Sci. Part A 1993, 31, 1755–1758. [Google Scholar] [CrossRef]
- Rhim, J.-W.; Park, H.-M.; Ha, C.-S. Bio-nanocomposites for food packaging applications. Prog. Polym. Sci. 2013, 38, 1629–1652. [Google Scholar] [CrossRef]
- Bikiaris, D.N. Nanocomposites of aliphatic polyesters: An overview of the effect of different nanofillers on enzymatic hydrolysis and biodegradation of polyesters. Polym. Degrad. Stab. 2013, 98, 1908–1928. [Google Scholar] [CrossRef]
- Vonk, C.G.; Kortleve, G. X-ray small-angle scattering of bulk polyethylene. Kolloid. Z. Z. Polym. 1967, 220, 19–24. [Google Scholar] [CrossRef]
- Vonk, C.G. A general computer program for the processing of small-angle X-ray scattering data. J. Appl. Crystallogr. 1975, 8, 340–341. [Google Scholar] [CrossRef]
- Tolunay, M.N.; Dawson, P.R.; Wang, P.P. Heating and bonding mechanisms in ultrasonic welding of thermoplastics. Polymer 1983, 23, 726–733. [Google Scholar] [CrossRef]
- Chatani, Y.; Okita, Y.; adokoro, H.; Yamashi, Y. Structural studies of polyesters. III. Crystal structure of poly-ε-caprolactone. Polymer J. 1970, 1, 555–562. [Google Scholar] [CrossRef]
- Hu, H.; Dorset, D.L. Crystal structure of poly(ε -caprolactone). Macromolecules 1990, 23, 4604–4607. [Google Scholar] [CrossRef]
- Iwata, T.; Doi, Y. Morphology and enzymatic degradation of poly(ε-caprolactone) single crystals: Does a polymer single crystal consist of micro-crystals? Polym. Int. 2002, 51, 852–858. [Google Scholar] [CrossRef]
- Li, L.L.; Li, B.; Hood, M.A.; Li, C.Y. Carbon nanotube induced polymer crystallization: The formation of nanohybrid shish-kebabs. Polymer 2009, 50, 953–965. [Google Scholar] [CrossRef]
- Hsiao, B.S.; Gardner, K.H.; Wu, D.Q.; Chu, B. Time-resolved X-ray study of poly(aryl ether ether ketone) crystallization and melting behaviour: 2. Melting. Polymer 1993, 34, 3986–4003. [Google Scholar] [CrossRef]
- Ikada, Y.; Jamshidi, K.; Tsuji, H.; Hyoan, S.H. Stereocomplex formation between enantiomeric poly(lactides). Macromolecules 1987, 20, 904–906. [Google Scholar] [CrossRef]
- Kruger, K.N.; Zachmann, H.G. Investigation of the melting behavior of poly(aryl ether ketones) by simultaneous measurements of SAXS and WAXS employing synchrotron radiation. Macromolecules 1993, 26, 5202–5208. [Google Scholar] [CrossRef]
- Hsiao, B.S.; Wang, Z.; Yeh, F.; Yang, G.; Sheth, K.C. Time-resolved X-ray studies of structure development in poly(butylene terephthalate) during isothermal crystallization. Polymer 1999, 40, 3515–3523. [Google Scholar] [CrossRef]
- Wurm, A.; Lellinger, D.; Minakov, A.A.; Skipa, T.; Pötschke, P.; Nicula, R.; Alig, I.; Schick, C. Crystallization of poly(ε-caprolactone)/MWCNT composites: A combined SAXS/WAXS, electrical and thermal conductivity study. Polymer 2014, 55, 2220–2232. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Amplitude (µm) | Force (N) | Time (s) | Mn (g/mol) | Mw (g/mol) | PI |
|---|---|---|---|---|---|---|
| PCL | - | - | - | 58,100 | 128,900 | 2.2 |
| PCL-1 1 | 43 | 2000 | 6 | 5900 | 49,200 | 8.4 |
| PCL-2 1 | 37 | 2000 | 6 | 47,000 | 120,100 | 2.6 |
| PCL-3 1 | 37 | 2500 | 6 | 56,700 | 127,300 | 2.2 |
| PCL-4 2 | 37 | 2500 | 8 | 58,100 | 128,900 | 2.2 |
| PCL-5 2 | 37 | 2500 | 7 | 57,500 | 128,100 | 2.2 |
| PCL6 1 | 37 | 2500 | 9 | 37,100 | 116,400 | 3.1 |
| PCL-7 2 | 37 | 2000 | 7 | 47,400 | 127,100 | 2.7 |
| MWCNT-5 2 | 37 | 2500 | 8 | 58,000 | 128,000 | 2.2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olmo, C.; Amestoy, H.; Casas, M.T.; Martínez, J.C.; Franco, L.; Sarasua, J.-R.; Puiggalí, J. Preparation of Nanocomposites of Poly(ε-caprolactone) and Multi-Walled Carbon Nanotubes by Ultrasound Micro-Molding. Influence of Nanotubes on Melting and Crystallization. Polymers 2017, 9, 322. https://doi.org/10.3390/polym9080322
Olmo C, Amestoy H, Casas MT, Martínez JC, Franco L, Sarasua J-R, Puiggalí J. Preparation of Nanocomposites of Poly(ε-caprolactone) and Multi-Walled Carbon Nanotubes by Ultrasound Micro-Molding. Influence of Nanotubes on Melting and Crystallization. Polymers. 2017; 9(8):322. https://doi.org/10.3390/polym9080322
Chicago/Turabian StyleOlmo, Cristian, Hegoi Amestoy, Maria Teresa Casas, Juan Carlos Martínez, Lourdes Franco, Jose-Ramon Sarasua, and Jordi Puiggalí. 2017. "Preparation of Nanocomposites of Poly(ε-caprolactone) and Multi-Walled Carbon Nanotubes by Ultrasound Micro-Molding. Influence of Nanotubes on Melting and Crystallization" Polymers 9, no. 8: 322. https://doi.org/10.3390/polym9080322
APA StyleOlmo, C., Amestoy, H., Casas, M. T., Martínez, J. C., Franco, L., Sarasua, J.-R., & Puiggalí, J. (2017). Preparation of Nanocomposites of Poly(ε-caprolactone) and Multi-Walled Carbon Nanotubes by Ultrasound Micro-Molding. Influence of Nanotubes on Melting and Crystallization. Polymers, 9(8), 322. https://doi.org/10.3390/polym9080322