
A Polyvinylpyrrolidone-Based Supersaturable Self-Emulsifying Drug Delivery System for Enhanced Dissolution of Cyclosporine A

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of CyA-Loaded Self-Emulsifying Formulations
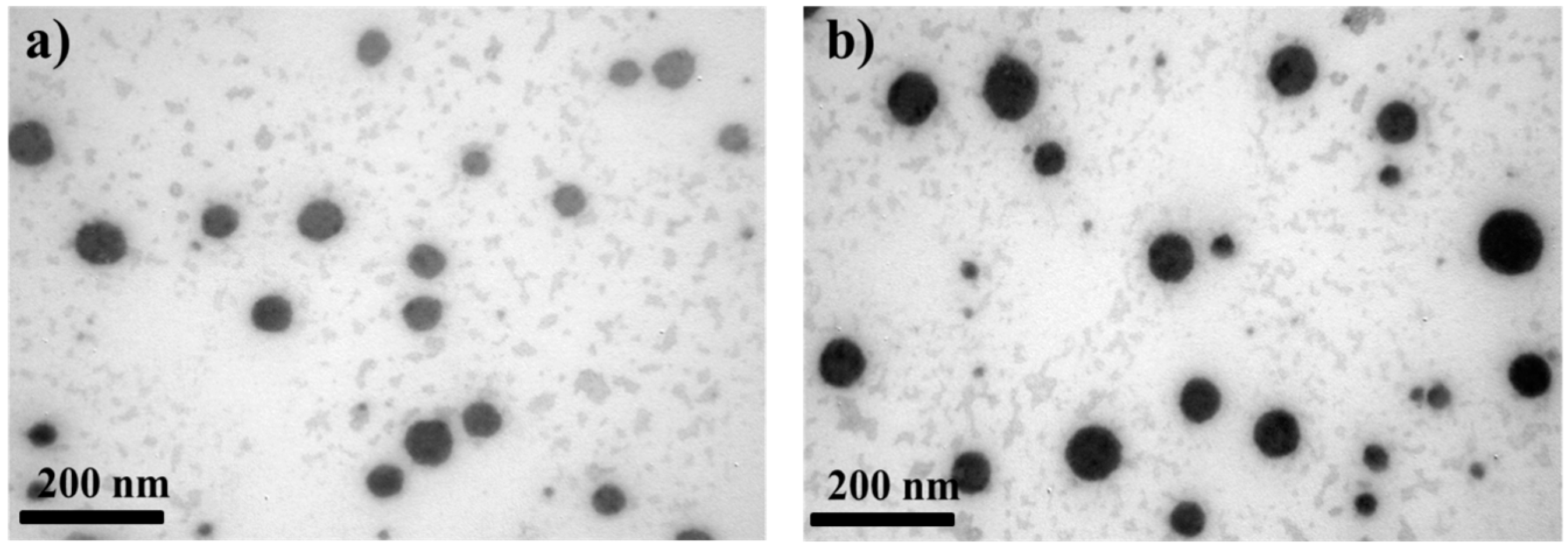
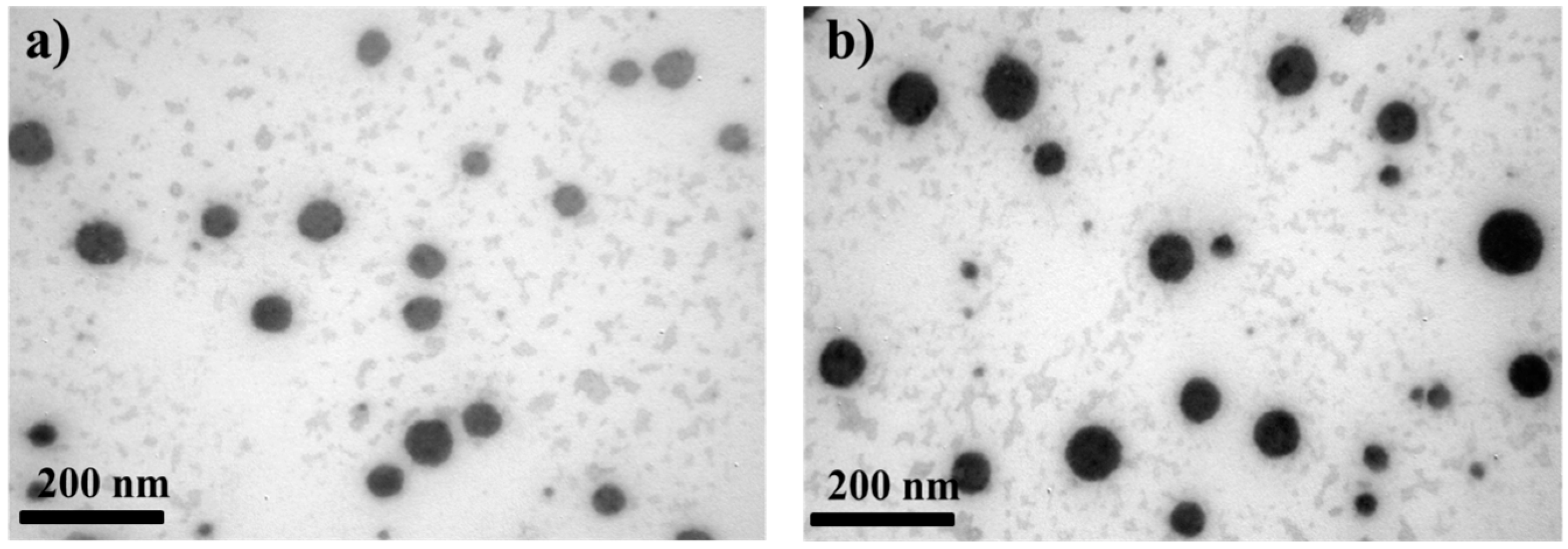
2.3. Morphology of CyA-Loaded Self-Emulsifying Formulations
2.4. Droplet Size and Zeta-Potential
2.5. Self-Emulsification Time
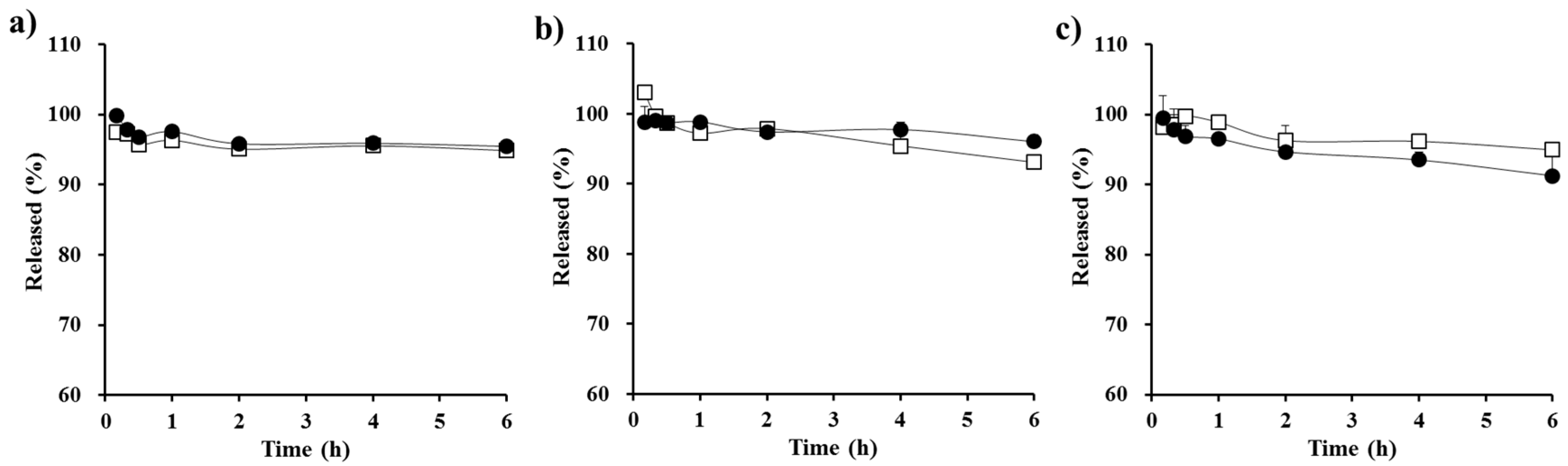
2.6. In Vitro Dialysis Test
2.7. HPLC Assay for CyA
2.8. Stability Test
3. Results and Discussion
3.1. Morphological and Physical Characteristics of Self-Emulsifying Formulations
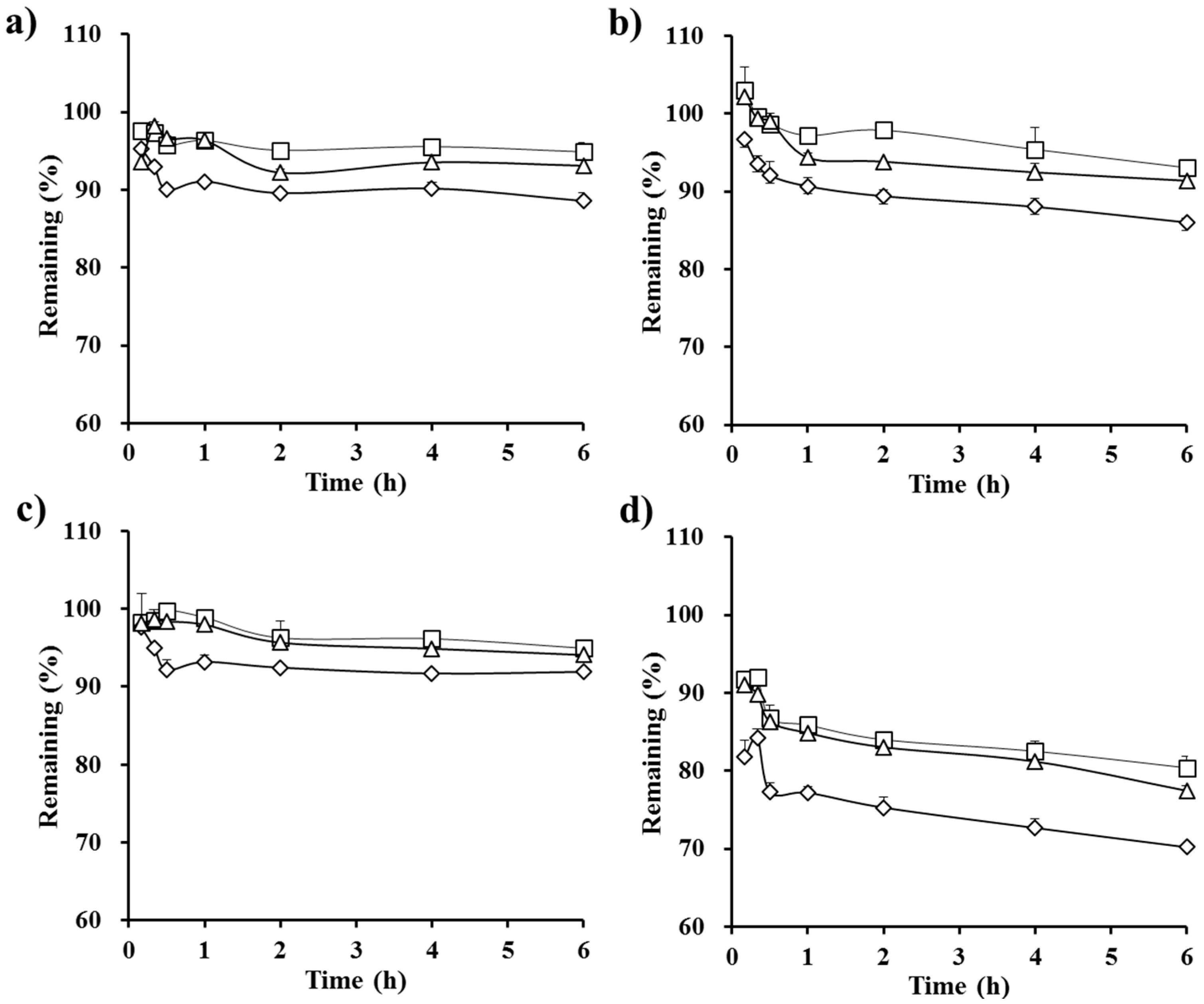
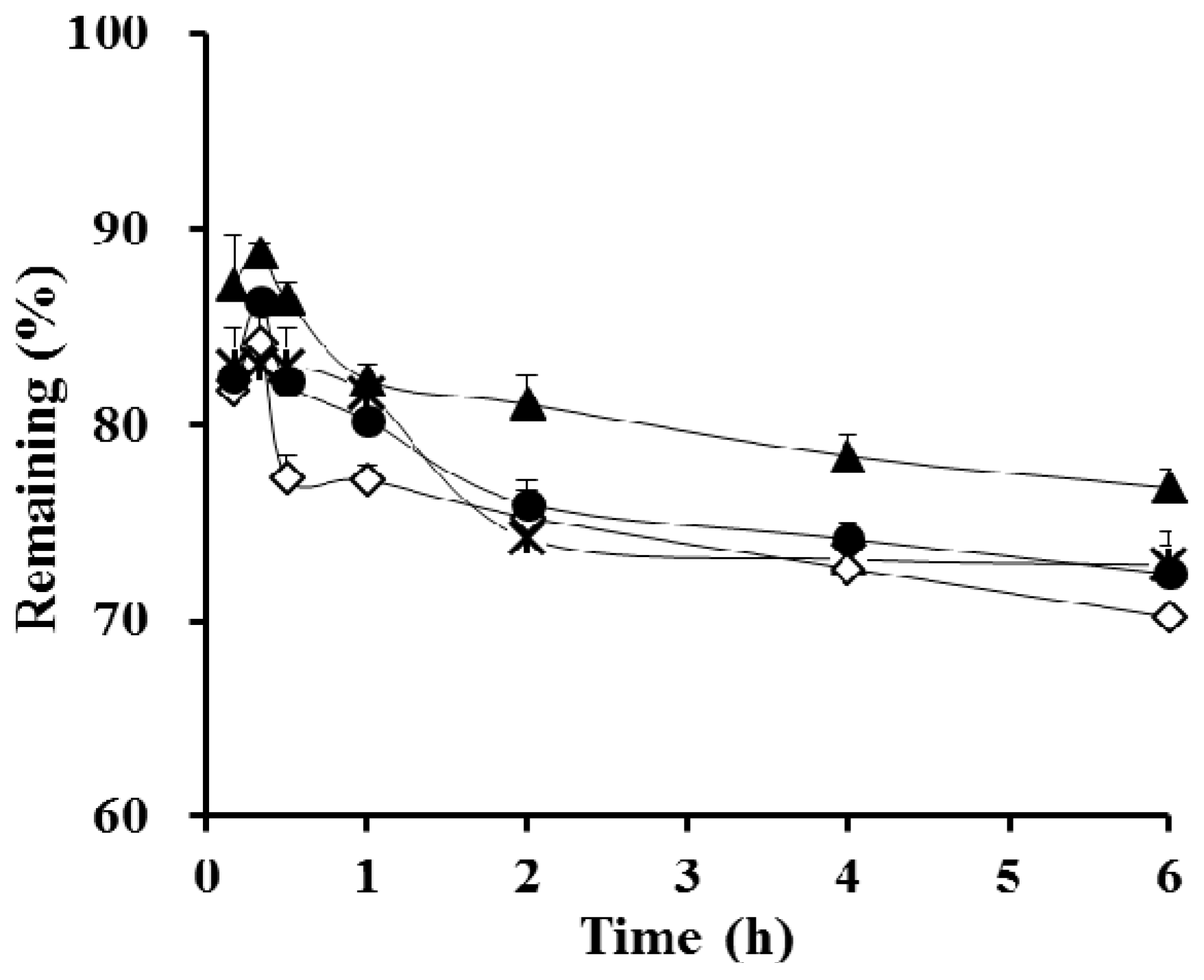
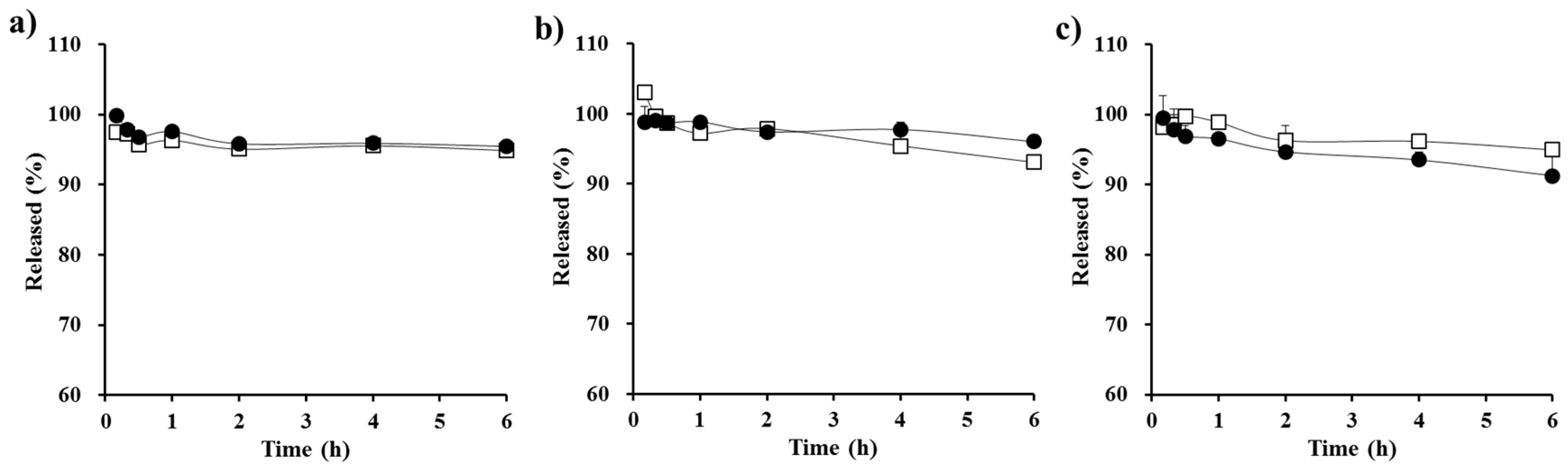
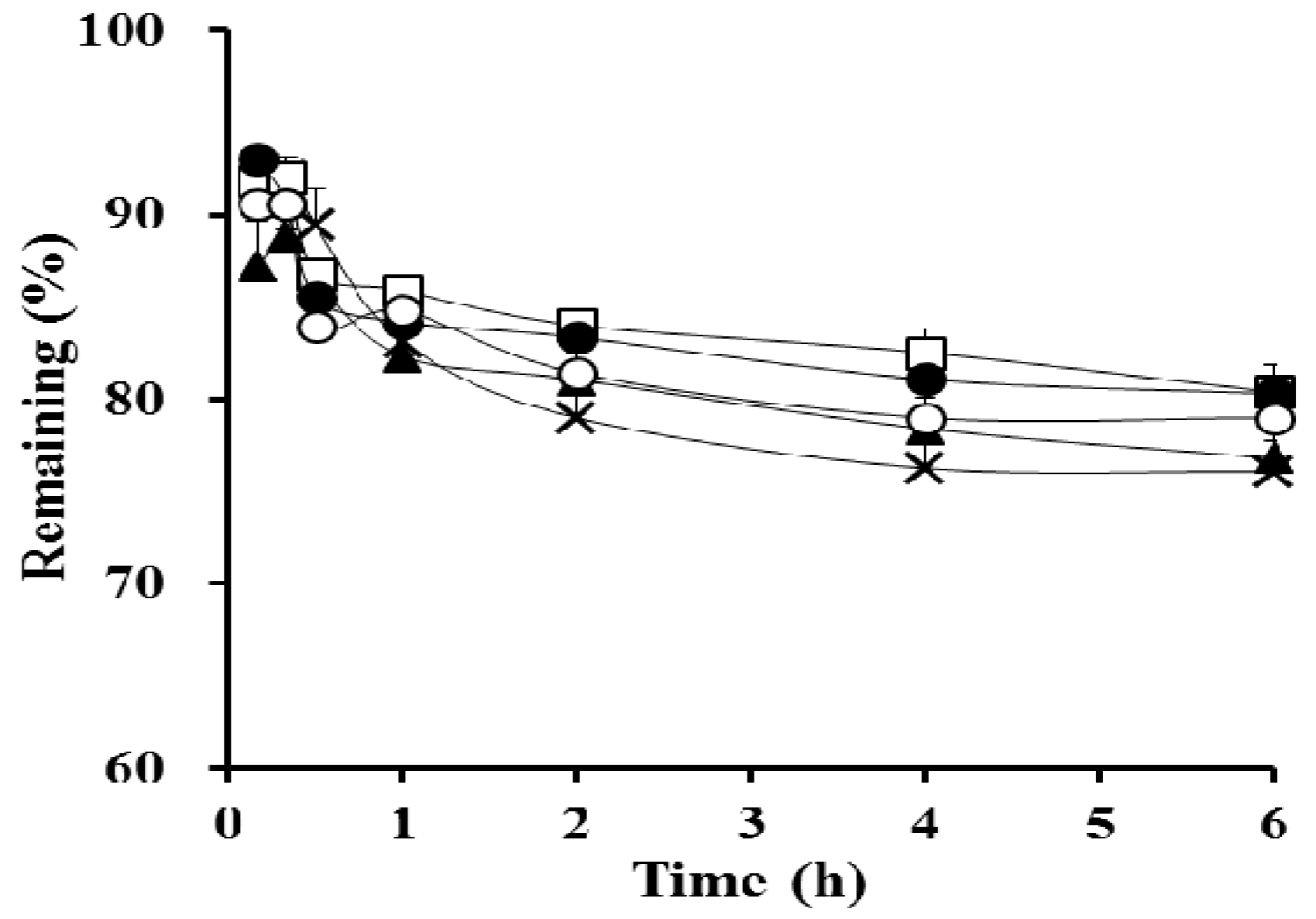
3.2. In Vitro Dialysis Test
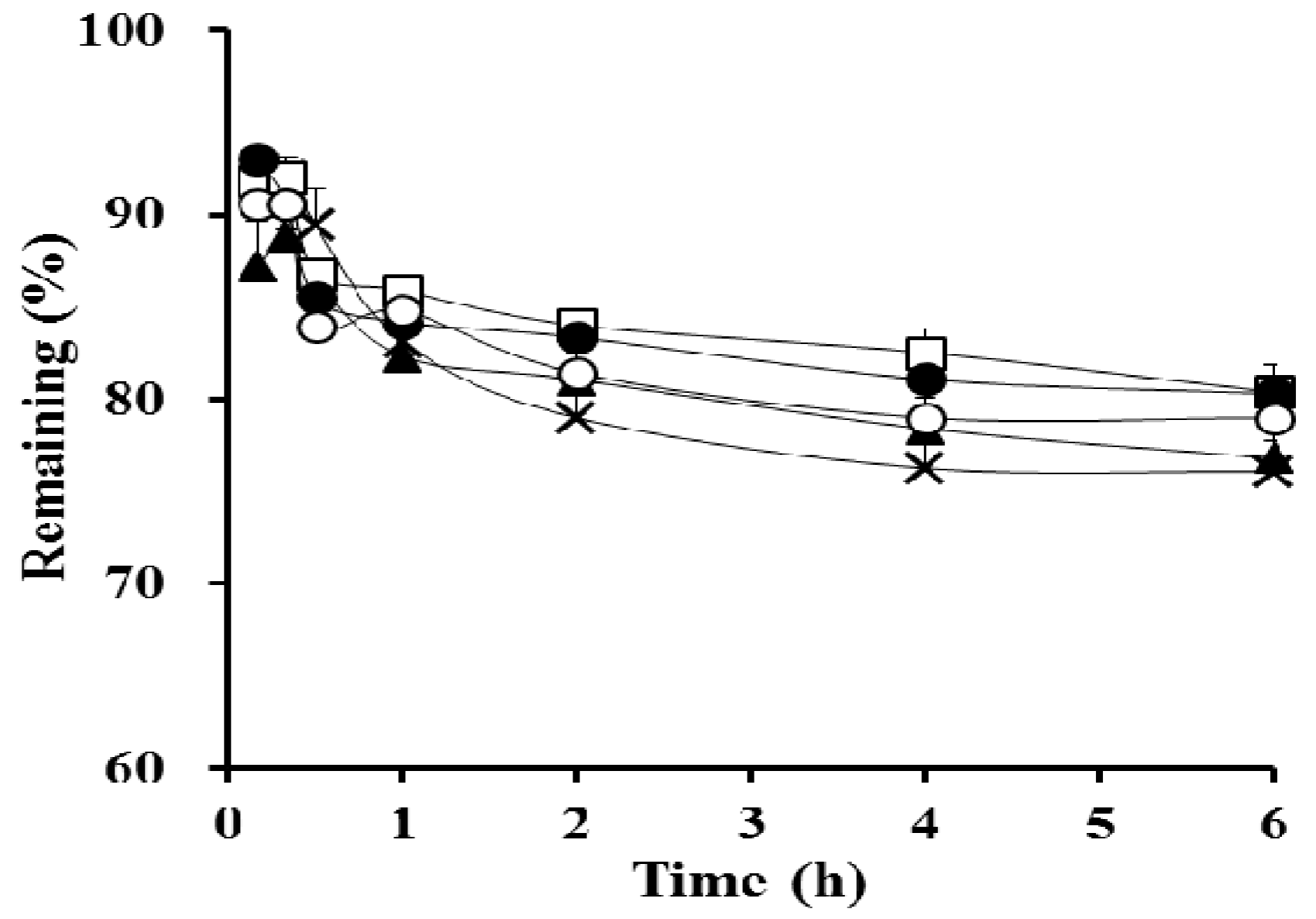
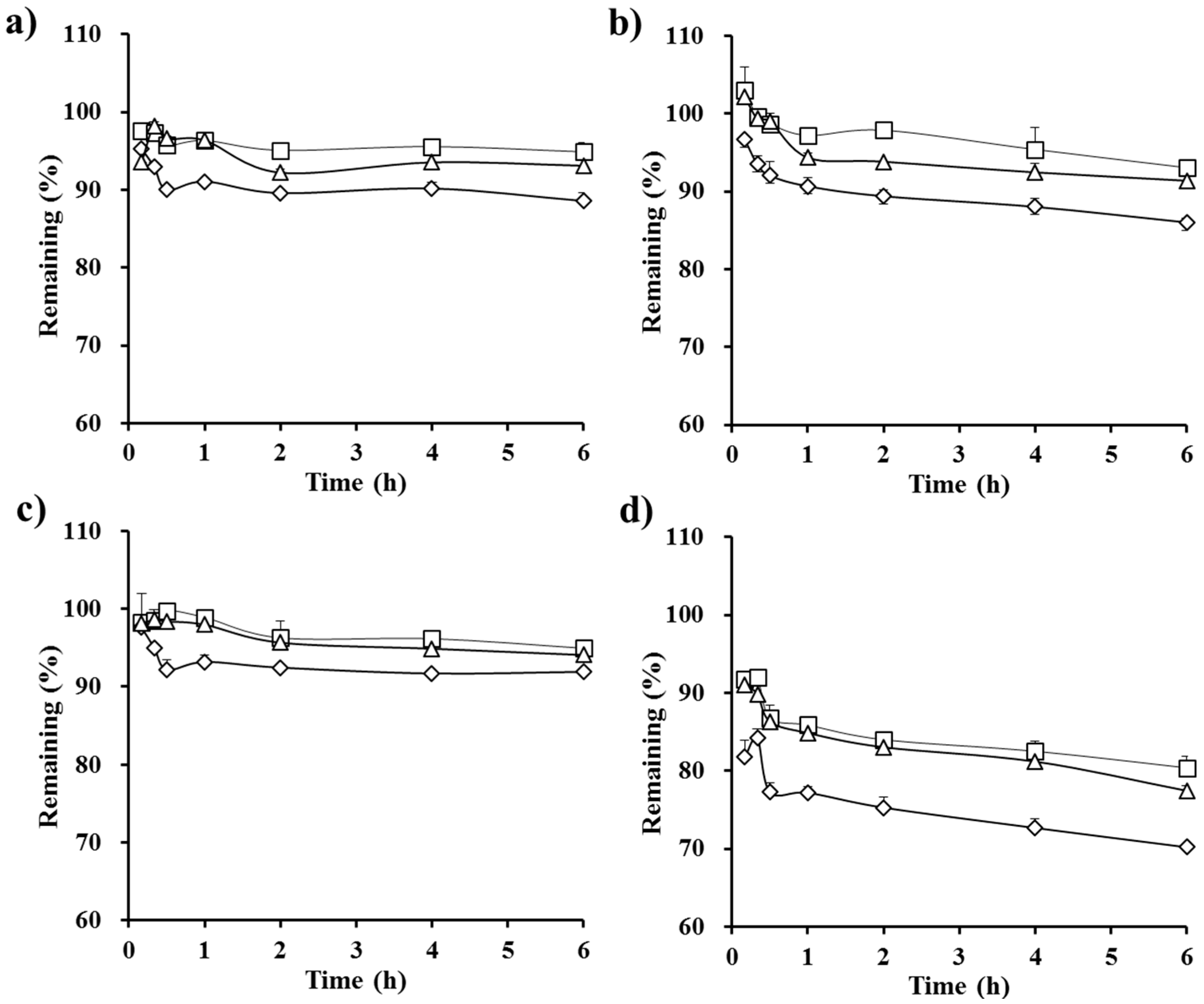
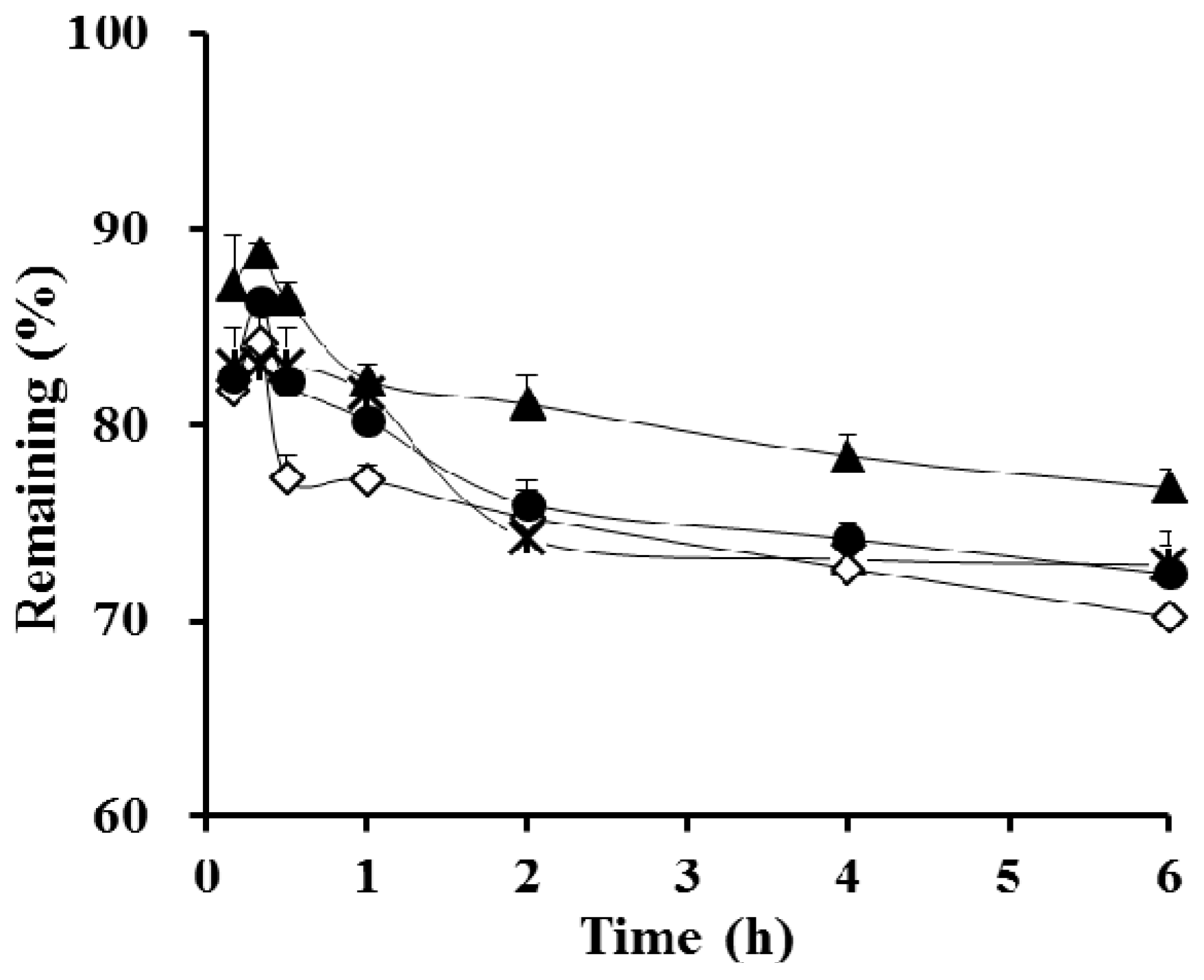
3.3. Stability Test
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Matzke, G.R.; Luke, D.R. Dialysis and renal transplant therapy. In Clinical Pharmacy and Therapeutics, 4th ed.; Herfindal, E.T., Gourley, D.R., Hart, L.L., Eds.; Williams and Wilkins: Baltimore, MD, USA, 1989. [Google Scholar]
- Luke, D.R.; Kasiske, B.L.; Matzke, G.R.; Awni, W.M.; Keane, W.F. Effect of cyclosporin on the isolated perfused rat kidney. Transplantation 1987, 43, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Noble, S.; Markham, A. Cyclosporin A review of the pharmacokinetic properties, clinical efficacy and tolerability of a microemulsion-based formulation (Neoral). Drugs 1995, 50, 924–941. [Google Scholar] [CrossRef] [PubMed]
- Ismailos, G.; Reppas, C.; Dressman, J.B.; Macheras, P. Unusual solubility behaviour of cyclosporin A in aqueous media. J. Pharm. Pharmacol. 1991, 43, 287–289. [Google Scholar] [CrossRef] [PubMed]
- USP Monographs of Neoral® Soft Gelatin Capsules. Available online: https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/neoral.pdf (accessed on 4 January 2017).
- Lawrence, M.J.; Rees, G.D. Microemulsion-based media as novel drug delivery systems. Adv. Drug Deliv. Rev. 2000, 45, 89–121. [Google Scholar] [CrossRef]
- Pouton, C.W. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000, 11, S93–S98. [Google Scholar] [CrossRef]
- Mueller, E.A.; Kovarik, J.M.; van Bree, J.B.; Grevel, J.; Luecker, P.W.; Kutz, K. Influence of a fat-rich meal on the pharmacokinetics of a new oral formulation of cyclosporine in a crossover comparison with the market formulation. Pharm. Res. 1994, 11, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Ritschel, W.A. Microemulsion technology in the reformulation of cyclosporine: The reason behind the pharmacokinetic properties of Neoral. Clin. Transplant. 1996, 10, 364–373. [Google Scholar] [PubMed]
- Cavanak, T.; Sucker, H. Formulation of dosage forms. Prog. Allergy 1986, 38, 65–72. [Google Scholar] [PubMed]
- Kiss, L.; Walter, F.R.; Bocsik, A.; Veszelka, S.; Ozsvári, B.; Puskás, L.G.; Szabó-Révész, P.; Deli, M.A. Kinetic analysis of the toxicity of pharmaceutical excipients Cremophor EL and RH40 on endothelial and epithelial cells. J. Pharm. Sci. 2013, 102, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating drug delivery systems: The answer to solubility-limited oral bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef] [PubMed]
- Van Speybroeck, M.; Mols, R.; Mellaerts, R.; Thi, T.D.; Martens, J.A.; Van Humbeeck, J.; Annaert, P.; Van den Mooter, G.; Augustijns, P. Combined use of ordered mesoporous silica and precipitation inhibitors for improved oral absorption of the poorly soluble weak base itraconazole. Eur. J. Pharm. Biopharm. 2010, 75, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Usui, F.; Maeda, K.; Kusai, A.; Nishimura, K.; Keiji, Y. Inhibitory effects of water soluble polymers on precipitation of RS-8359. Int. J. Pharm. 1997, 154, 59–66. [Google Scholar] [CrossRef]
- Loftsson, T.; Friðriksdóttir, H.; Guðmundsdóttir, T.K. The effect of water-soluble polymers on aqueous solubility of drugs. Int. J. Pharm. 1996, 127, 293–296. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, W.; Jin, Y.; Quan, D.Q. Studies on preparation of carbamazepine (CBZ) supersaturatable self-microemulsifying (S-SMEDDS) formulation and relative bioavailability in beagle dogs. Pharm. Dev. Technol. 2011, 16, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Tajarobi, F.; Larsson, A.; Matic, H.; Abrahmsen-Alami, S. The influence of crystallization inhibition of HPMC and HPMCAS on model substance dissolution and release in swellable matrix tablets. Eur. J. Pharm. Biopharm. 2011, 78, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.L.; Trividic, A.; Davis, A.F.; Hadgraft, J. Crystallization of hydrocortisone acetate: Influence of polymers. Int. J. Pharm. 2001, 212, 213–221. [Google Scholar] [CrossRef]
- Zimmermann, A.; Millqvist-Fureby, A.; Elema, M.R.; Hansen, T.; Mullertz, A.; Hovgaard, L. Adsorption of pharmaceutical excipients onto microcrystals of siramesine hydrochloride: Effects on physicochemical properties. Eur. J. Pharm. Biopharm. 2009, 71, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Rush, B.D.; Pfund, W.P.; Huang, T.; Bauer, J.M.; Morozowich, W.; Kuo, M.S.; Hageman, M.J. Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J. Pharm. Sci. 2003, 92, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Guyton, M.E.; Huang, T.; Bauer, J.M.; Stefanski, K.J.; Lu, Q. Enhanced oral bioavailability of a poorly water soluble drug PNU-91325 by supersaturatable formulations. Drug Dev. Ind. Pharm. 2004, 30, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Akrami, A.; Alvarez, F.; Hu, J.; Li, L.; Ma, C.; Surapaneni, S. Characterization and optimization of AMG 517 supersaturable self-emulsifying drug delivery system (S-SEDDS) for improved oral absorption. J. Pharm. Sci. 2009, 98, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Klyashchitsky, B.A.; Owen, A.J. Drug delivery systems for cyclosporine: Achievements and complications. J. Drug Target. 1998, 5, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Vertzoni, M.; Dressman, J.; Butler, J.; Hempenstall, J.; Reppas, C. Simulation of fasting gastric conditions and its importance for the in vivo dissolution of lipophilic compounds. Eur. J. Pharm. Biopharm. 2005, 60, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Galia, E. Physiologically Based Dissolution Tests. Ph.D. Thesis, Johann Wolfgang Goethe University, Frankfurt, Germany, 1999. [Google Scholar]
- Macheras, P.; Koupparis, M.; Tsaprounis, C. Drug dissolution studies in milk using the automated flow injection serial dynamic dialysis technique. Int. J. Pharm. 1986, 33, 125–136. [Google Scholar] [CrossRef]
- Galia, E.; Nicolaides, E.; Horter, D.; Lobenberg, R.; Reppas, C.; Dressman, J.B. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm. Res. 1998, 15, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution media simulating conditions in the proximal human gastrointestinal tract: An update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.H.; Carvajal, M.T.; Patel, C.I.; Infeld, M.H.; Malick, A.W. Self-emulsifying drug delivery systems (SEDDS) with polyglycolyzed glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. Int. J. Pharm. 1994, 106, 15–23. [Google Scholar] [CrossRef]
- Tarr, B.D.; Yalkowsky, S.H. Enhanced intestinal absorption of cyclosporine in rats through the reduction of emulsion droplet size. Pharm. Res. 1989, 6, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.; Holm, P. Porous magnesium aluminometasilicate tablets as carrier of a cyclosporine self-emulsifying formulation. AAPS PharmSciTech 2009, 10, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Onoue, S.; Sato, H.; Ogawa, K.; Kawabata, Y.; Mizumoto, T.; Yuminoki, K.; Hashimoto, N.; Yamada, S. Improved dissolution and pharmacokinetic behavior of cyclosporine A using high-energy amorphous solid dispersion approach. Int. J. Pharm. 2010, 399, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Q.; Liu, Y.; Zhao, J.H.; Wang, L.; Feng, N.P. Improved oral bioavailability of poorly water-soluble indirubin by a supersaturatable self-microemulsifying drug delivery system. Int. J. Nanomed. 2012, 7, 1115–1125. [Google Scholar]
- Balani, P.N.; Wong, S.Y.; Ng, W.K.; Widjaja, E.; Tan, R.B.H.; Chan, S.Y. Influence of polymer content on stabilizing milled amorphous salbutamol sulphate. Int. J. Pharm. 2010, 391, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Anby, M.U.; Williams, H.D.; McIntosh, M.; Benameur, H.; Edwards, G.A.; Pouton, C.W.; Porter, C.J. Lipid digestion as a trigger for supersaturation: Evaluation of the impact of supersaturation stabilization on the in vitro and in vivo performance of self-emulsifying drug delivery systems. Mol. Pharm. 2012, 9, 2063–2079. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 |
|---|---|---|---|---|---|---|---|---|---|
| Compositions | |||||||||
| CyA (mg) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Maisine 35-1 (μL) | 320 | 240 | 160 | 160 | 160 | 160 | 160 | 160 | 160 |
| Kolliphor RH40 (μL) | 380 | 285 | 190 | 190 | 190 | 190 | 190 | 190 | 190 |
| Ethanol (μL) | 100 | 75 | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| Propylene glycol (μL) | 100 | 75 | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| HPC (mg) | 15 | ||||||||
| Kollidone VA64 | 15 | ||||||||
| PVP (mg) | 15 | 20 | 30 | 40 | |||||
| Physical characteristics | |||||||||
| Size (nm) | 39.4 | 36.7 | 102.5 * | 123.0 * | 120.6 * | 113.4 * | 104.2 * | 114.1 * | 120.1 * |
| Polydispersity index | 0.33 | 0.26 | 0.28 | 0.28 | 0.29 | 0.32 | 0.35 | 0.38 | 0.25 |
| Zeta potential (mV) | −5.1 | −4.1 | −5.4 | −5.5 | −3.8 | −4.2 | −5.6 | −3.8 | −4.4 |
| Emulsification time (min) | 8.5 | 7.4 | 6.2 * | 15.5 * | 6.1 * | 6.2 * | 5.4 * | 6.5 * | 5.8 * |
| Parameters | Immediate after Preparation | After 6 Months Storage |
|---|---|---|
| Droplet size (nm) a | 114.1 | 126.3 |
| Zeta potential (mV) a | −4.2 | −4.5 |
| Emulsification time (min) a | 6.5 | 5.8 |
| Remaining at 10 min (%) b | 93.0 ± 0.5 | 91.8 ± 1.5 |
| Remaining at 6 h (%) b | 80.3 ± 1.5 | 81.4 ± 5.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.R.; Ho, M.J.; Choi, Y.W.; Kang, M.J. A Polyvinylpyrrolidone-Based Supersaturable Self-Emulsifying Drug Delivery System for Enhanced Dissolution of Cyclosporine A. Polymers 2017, 9, 124. https://doi.org/10.3390/polym9040124
Lee DR, Ho MJ, Choi YW, Kang MJ. A Polyvinylpyrrolidone-Based Supersaturable Self-Emulsifying Drug Delivery System for Enhanced Dissolution of Cyclosporine A. Polymers. 2017; 9(4):124. https://doi.org/10.3390/polym9040124
Chicago/Turabian StyleLee, Dae Ro, Myoung Jin Ho, Young Wook Choi, and Myung Joo Kang. 2017. "A Polyvinylpyrrolidone-Based Supersaturable Self-Emulsifying Drug Delivery System for Enhanced Dissolution of Cyclosporine A" Polymers 9, no. 4: 124. https://doi.org/10.3390/polym9040124
APA StyleLee, D. R., Ho, M. J., Choi, Y. W., & Kang, M. J. (2017). A Polyvinylpyrrolidone-Based Supersaturable Self-Emulsifying Drug Delivery System for Enhanced Dissolution of Cyclosporine A. Polymers, 9(4), 124. https://doi.org/10.3390/polym9040124