
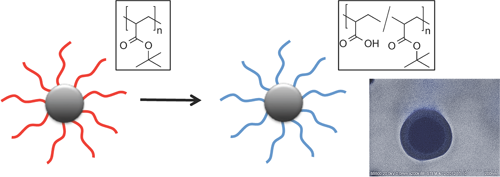
Stable Poly(methacrylic acid) Brush Decorated Silica Nano-Particles by ARGET ATRP for Bioconjugation

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Chemicals
2.2. Methods
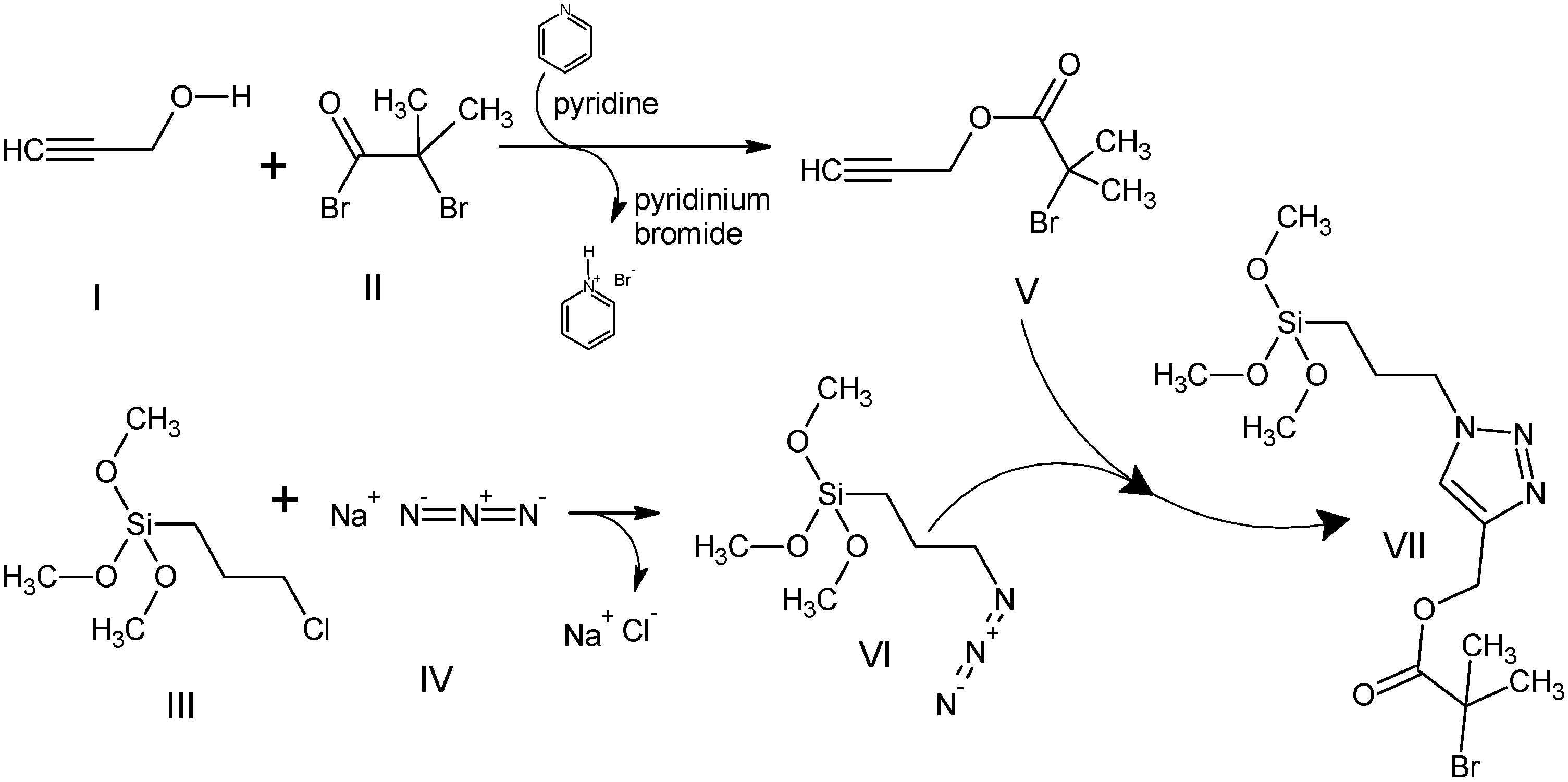
2.3. Synthesis of Si-NP (Diameter 200 nm)
2.4. Decoration of Silica Nano-Particles with ATRP Initiator VII
2.5. Grafting of Tert-Butyl Methacrylate from ATRP Initiator Decorated Silica Nano-Particles
2.6. Reaction Kinetics and Degrafting of Polymer from Si-NP
2.7. Deprotection of Poly(tert-Butyl Methacrylate) Grafted Si-NP
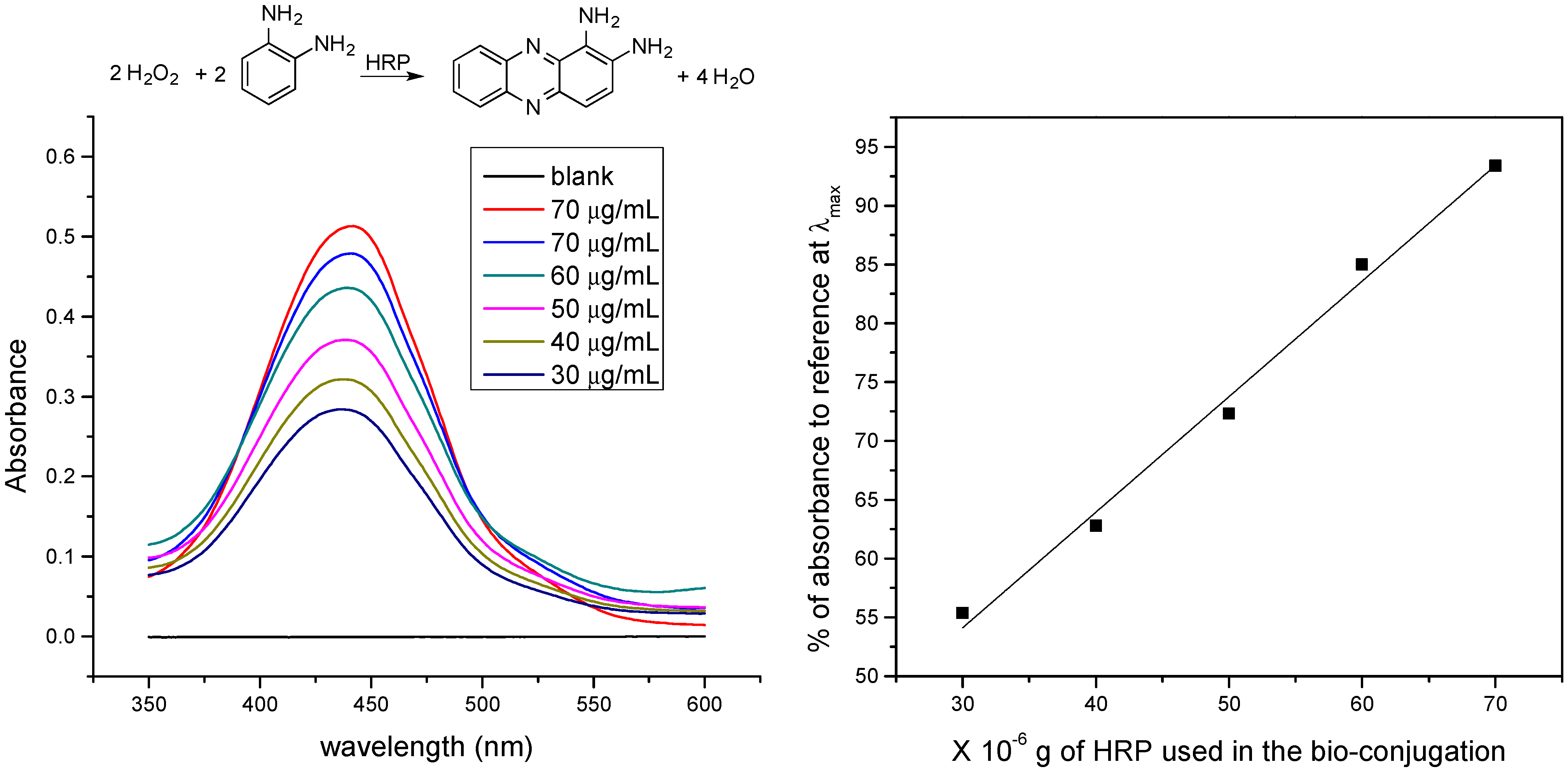
2.8. Bioconjugation with HRP
2.9. Colorimetric Test
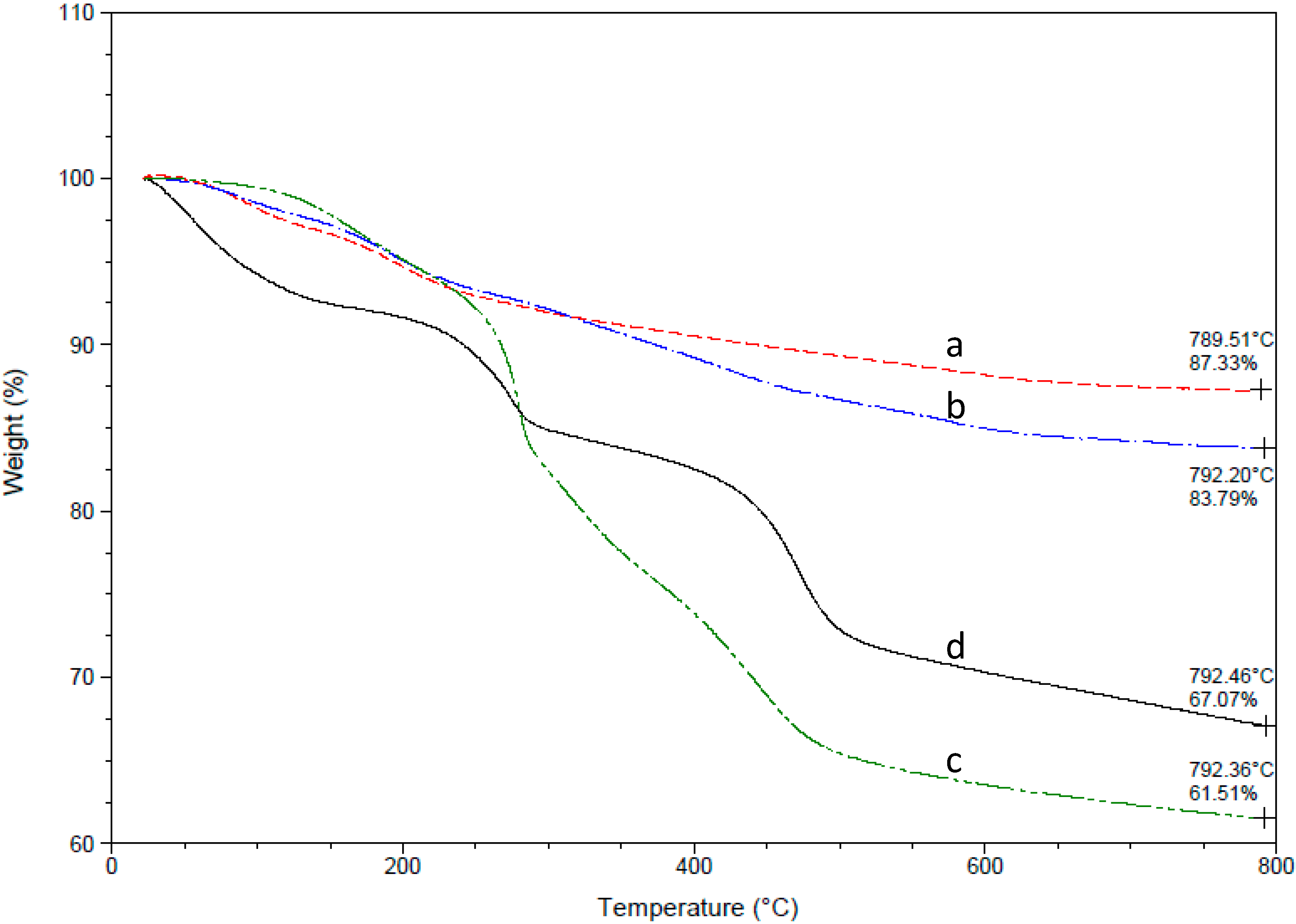
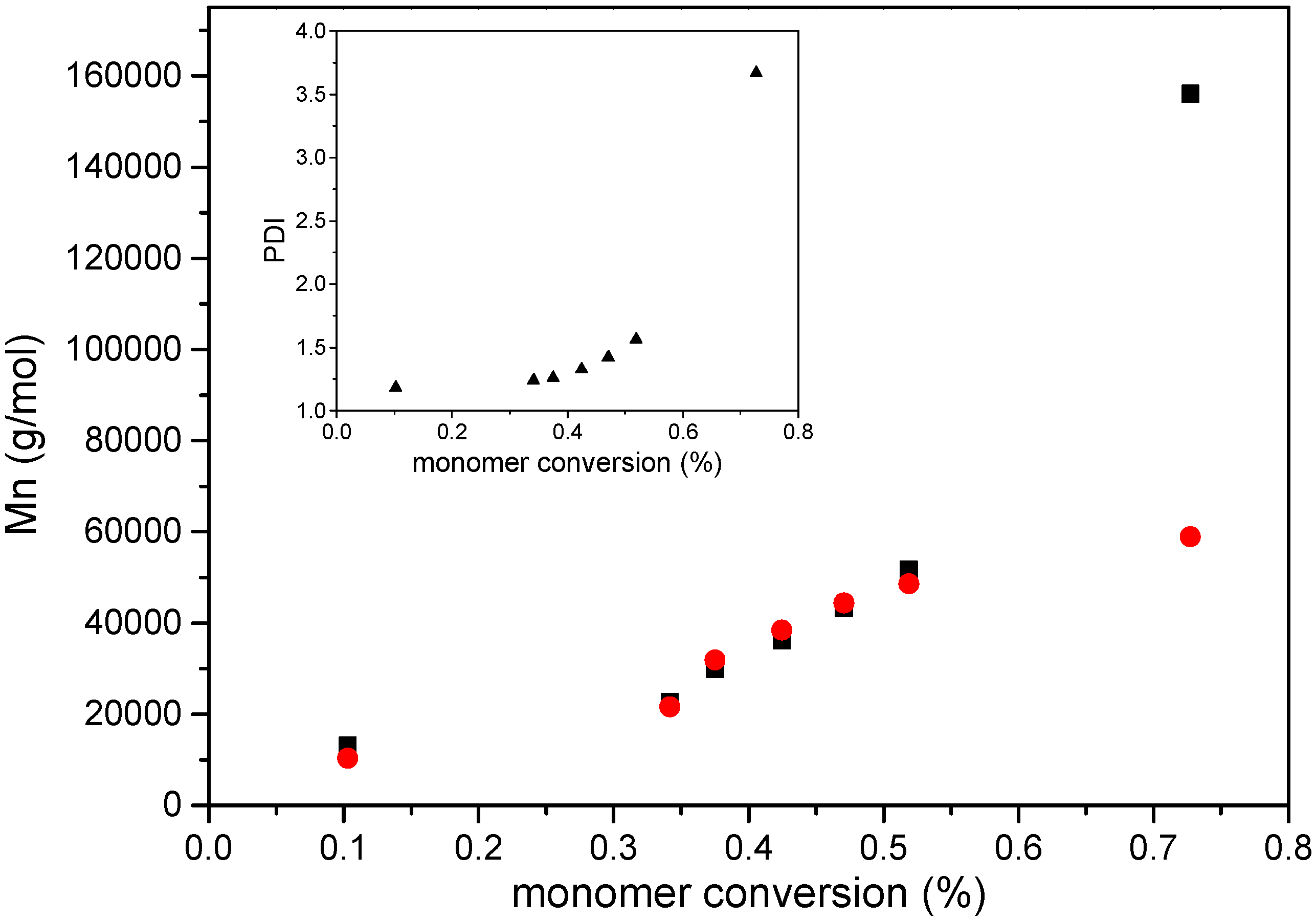
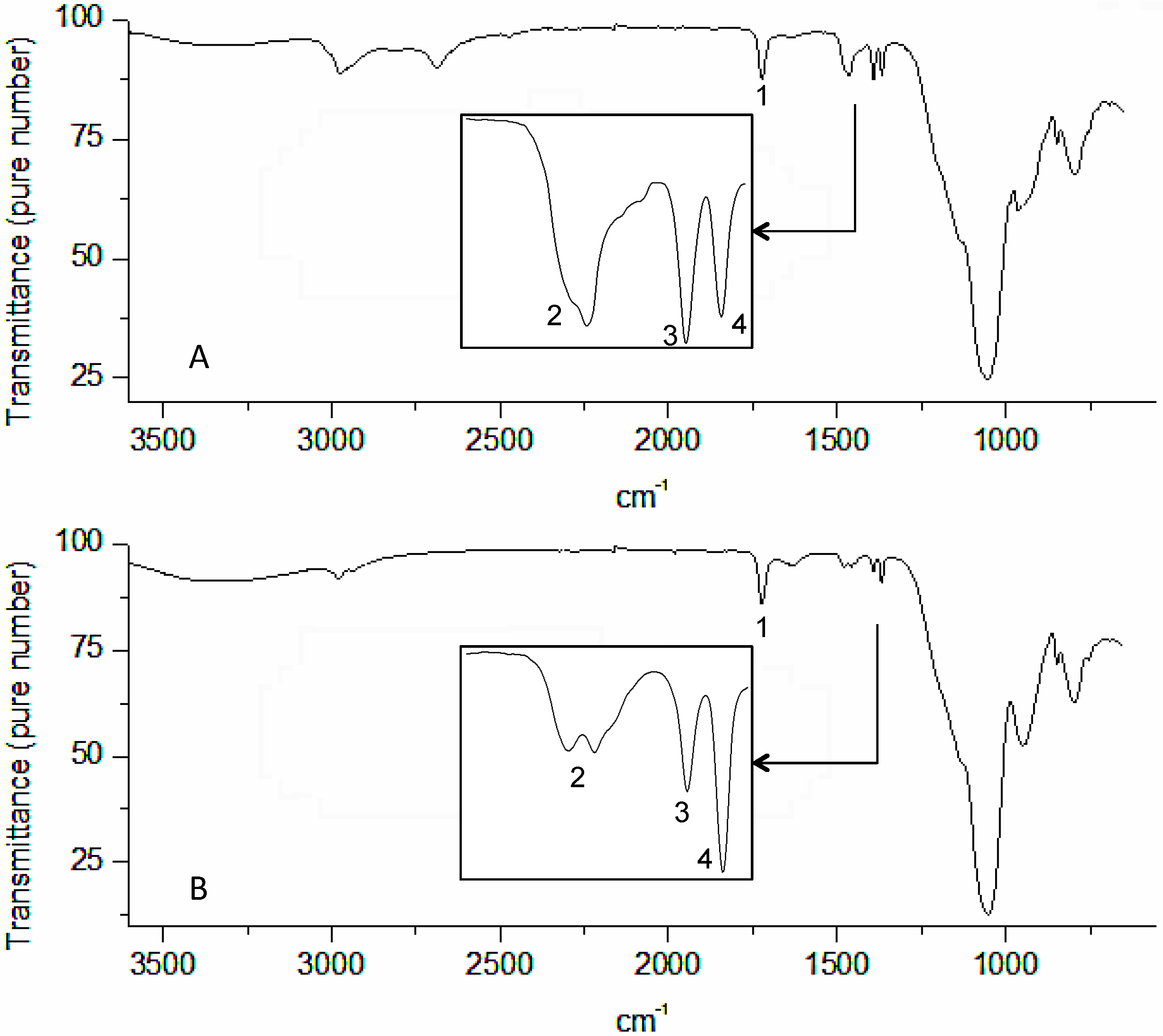
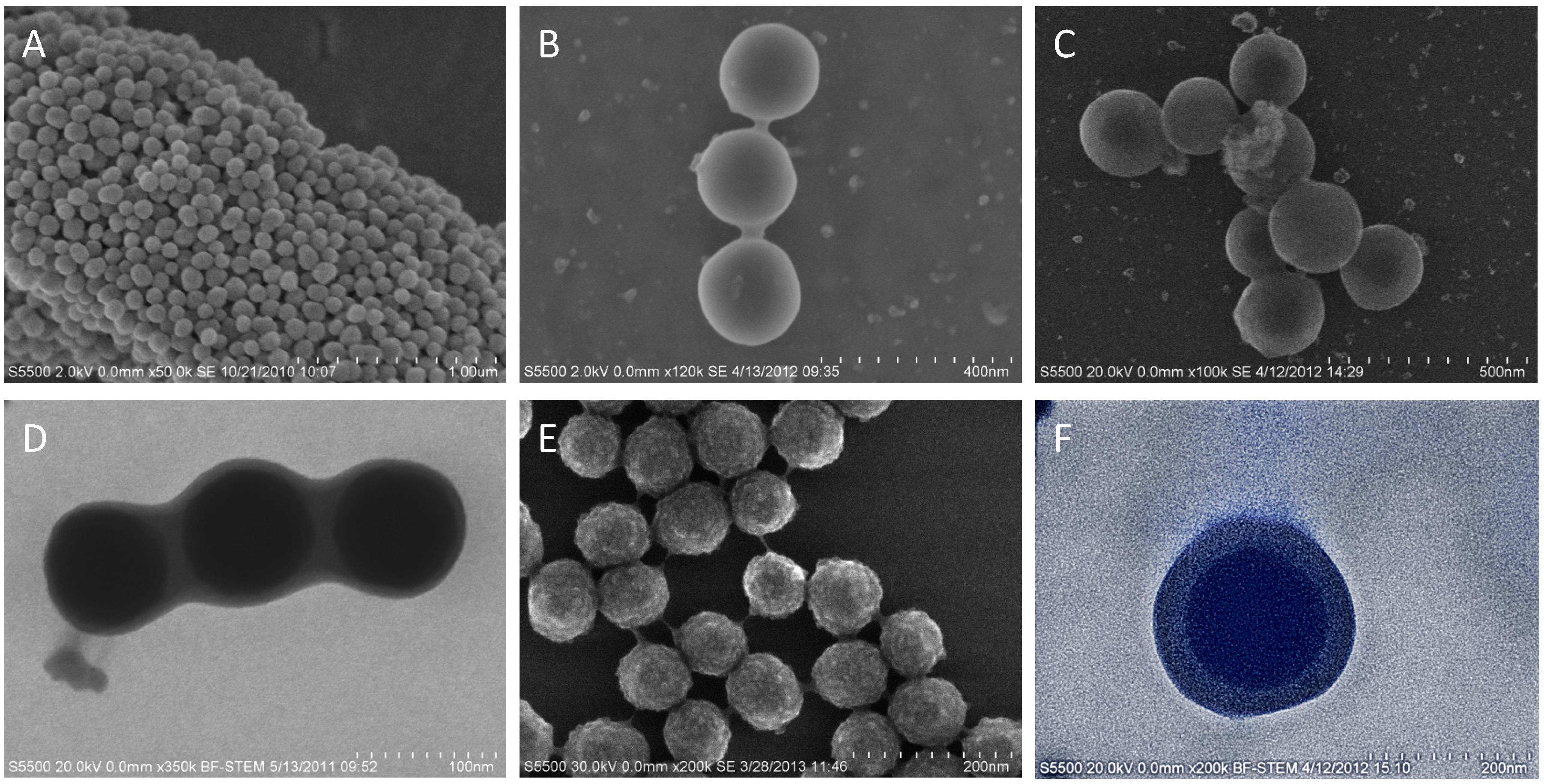
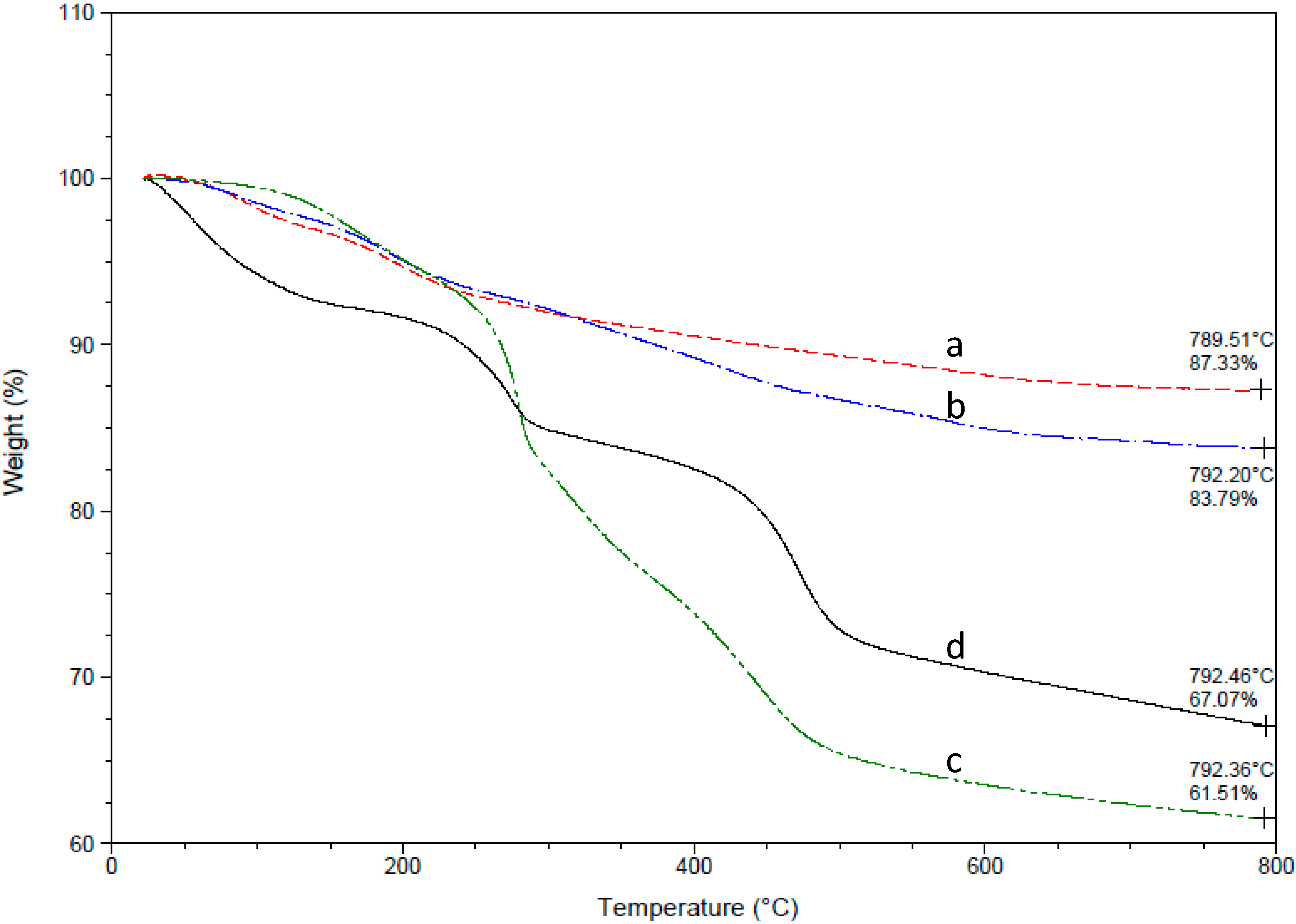
3. Results and Discussion
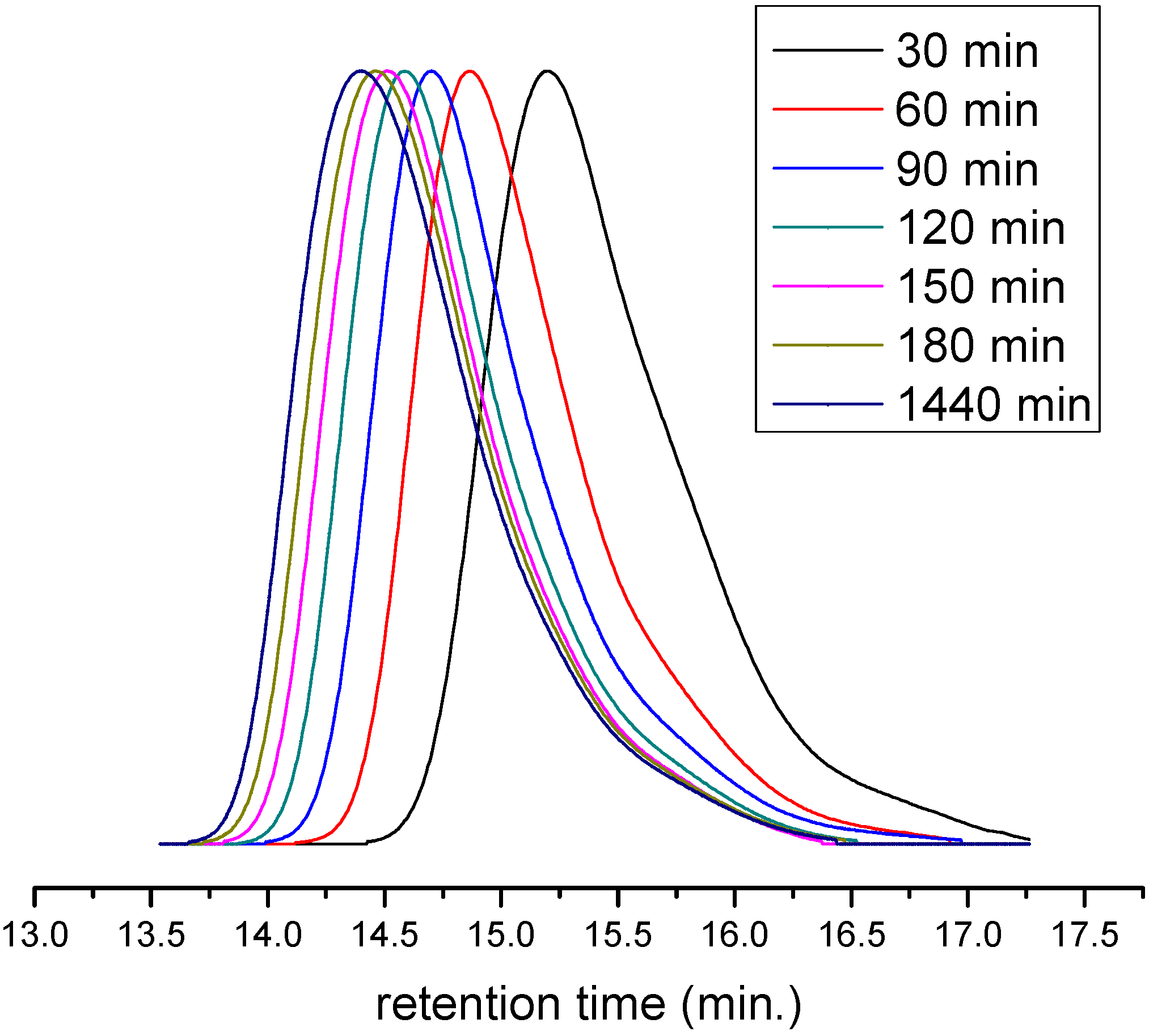
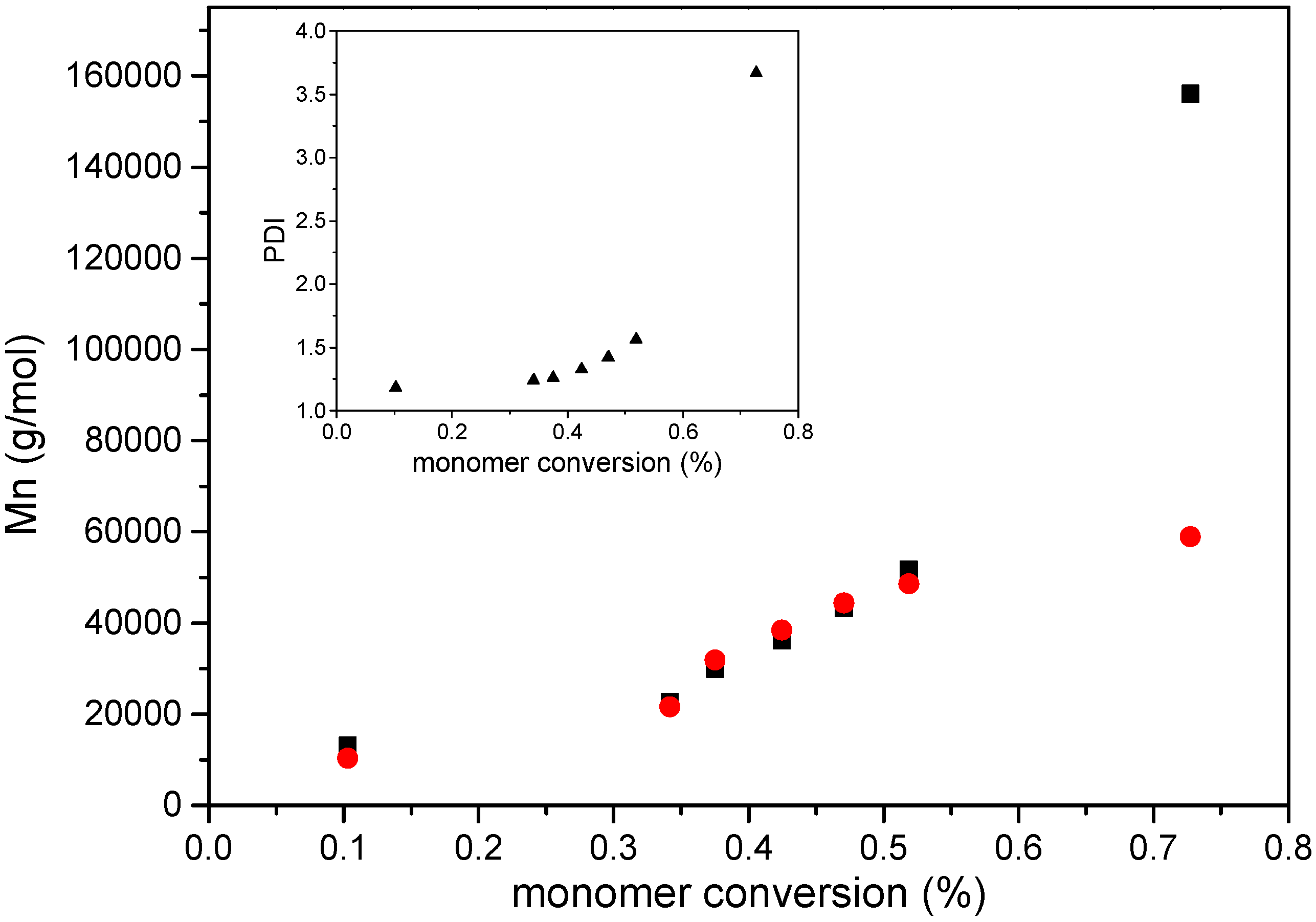
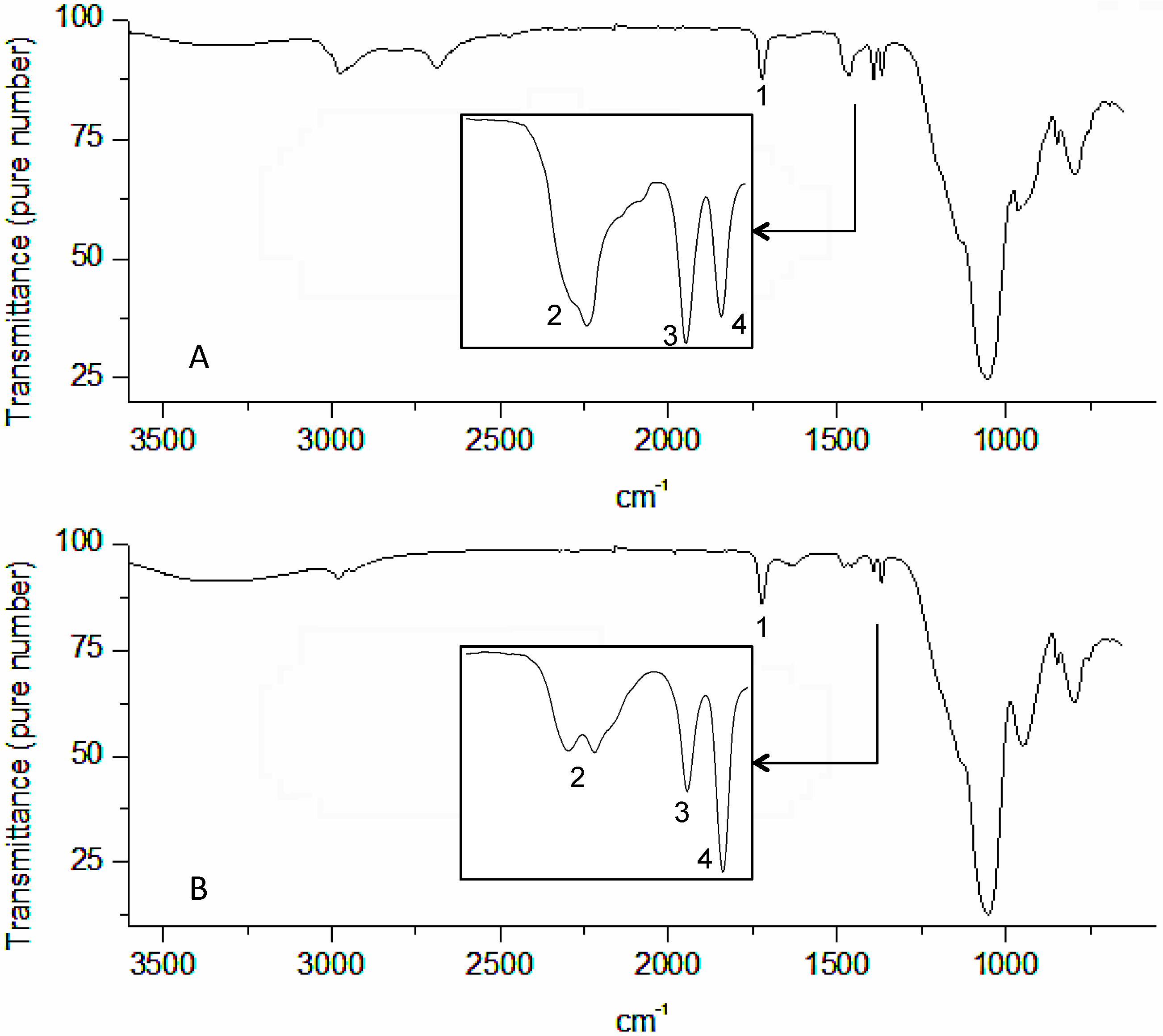
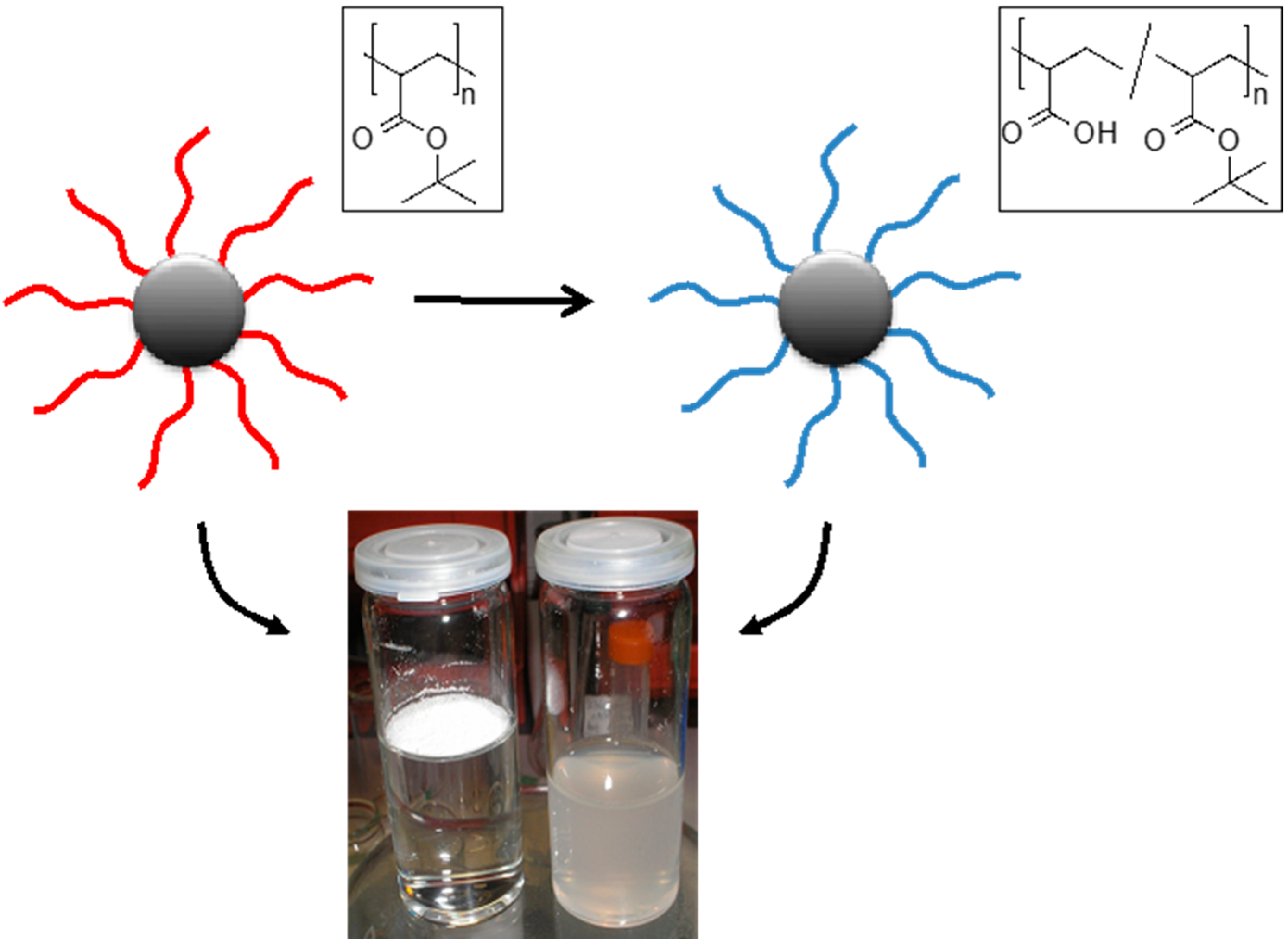
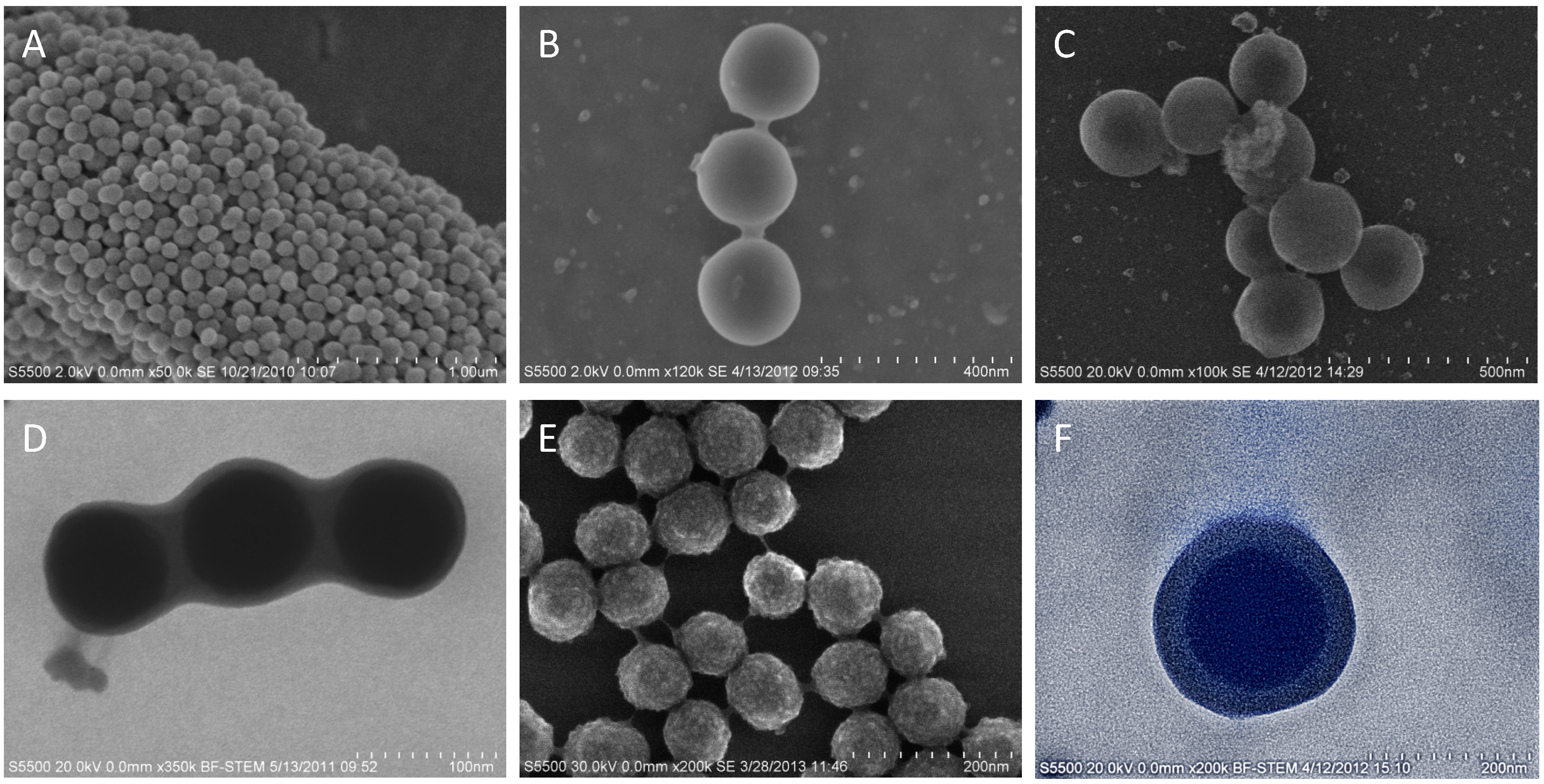
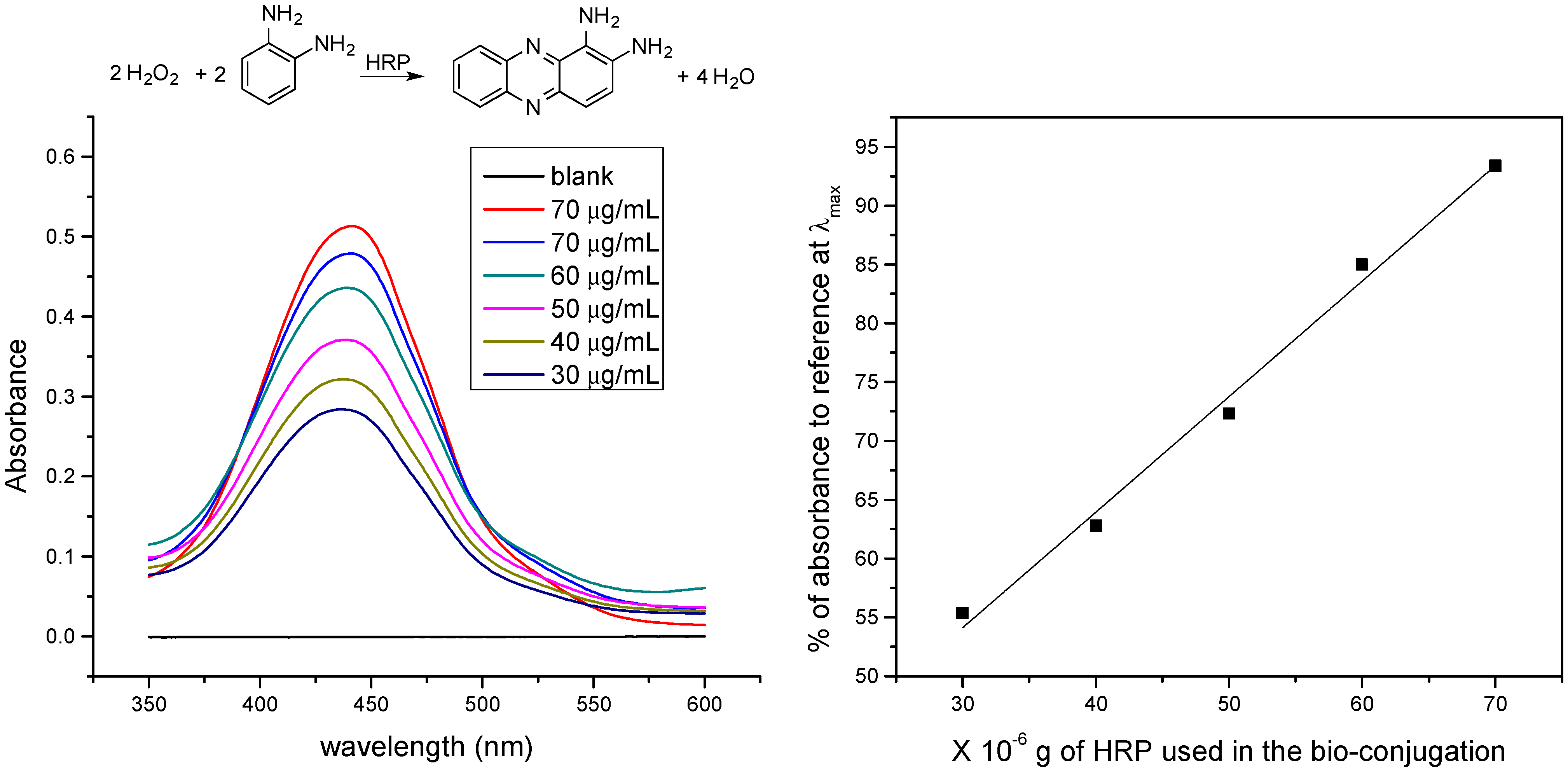
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jiang, T.; Mo, R.; Bellotti, A.; Zhou, J.; Gu, Z. Gel–liposome-mediated co-delivery of anticancer membrane-associated proteins and small-molecule drugs for enhanced therapeutic efficacy. Adv. Funct. Mater. 2014, 24, 2295–2304. [Google Scholar] [CrossRef]
- Hood, J.L.; Jallouck, A.P.; Campbell, N.; Ratner, L.; Wickline, S.A. Cytolytic nanoparticles attenuate HIV-1 infectivity. Antivir. Ther. 2013, 18, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Thaxton, C.S.; Elghanian, E.; Thomas, A.D.; Stoeva, S.I.; Lee, J.-S.; Smith, N.D.; Schaeffer, A.J.; Klocker, H.; Horninger, W.; Bartsch, G.; et al. Nanoparticle-based bio-barcode assay redefines “undetectable” PSA and biochemical recurrence after radical prostatectomy. Proc. Natl. Acad. Sci. USA 2009, 106, 18437–18442. [Google Scholar] [CrossRef] [PubMed]
- Getts, D.R.; Martin, A.J.; McCarthy, D.P.; Terry, R.L.; Hunter, Z.N.; Yap, W.T.; Getts, M.T.; Pleiss, M.; Luo, X.R.; King, N.J.C.; et al. Microparticles bearing encephalitogenic peptides induce t-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat. Biotechnol. 2012, 30, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Y.; Xiang, R.; Ciuparu, D.; Pfefferle, L.D.; Horwath, C.; Wilkins, J.A. Incorporation of single-wall carbon nanotubes into an organic polymer monolithic stationary phase for µ-HPLC and capillary electrochromatography. Anal. Chem. 2005, 77, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Chambers, S.D.; Holcombe, T.W.; Svec, F.; Frechet, J.M.J. Porous polymer monoliths functionalized through copolymerization of a C60 fullerene-containing methacrylate monomer for highly efficient separations of small molecules. Anal. Chem. 2011, 83, 9478–9484. [Google Scholar] [CrossRef] [PubMed]
- Stöber, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid. Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Nyffenegger, R.; Quellet, C.; Ricka, J. Synthesis of fluorescent, monodisperse, colloidal silica particles. J. Colloid Interface Sci. 1993, 159, 150–157. [Google Scholar] [CrossRef]
- Xu, J.Q.; Sun, L.; Li, J.; Liang, J.L.; Zhang, H.M.; Yang, W.S. FITC and Ru(phen)32+ co-doped silica particles as visualized ratiometric ph indicator. Nanoscale Res. Lett. 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Bin, Z.; Brittain, W.J. Polymer brushes: Surface-immobilized macromolecules. Prog. Polym. Sci. 2000, 25, 677–710. [Google Scholar]
- Edmondson, S.; Steve, E.; Osborne, V.L.; Huck, W.T.S. Polymer brushes via surface-initiated polymerizations. Chem. Soc. Rev. 2004, 33, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Audouin, F.; Larragy, R.; Fox, M.; O’Connor, B.; Heise, A. Protein immobilization onto poly(acrylic acid) functional macroporous polyhipe obtained by surface-initiated ARGET ATRP. Biomacromolecules 2012, 13, 3787–3794. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.F.; Ruckenstein, E. Water-soluble poly(acrylic acid) grafted luminescent silicon nanoparticles and their use as fluorescent biological staining labels. Nano Lett. 2004, 4, 1463–1467. [Google Scholar] [CrossRef]
- Chunzhao, L.; Benicewicz, B.C. Synthesis of well-defined polymer brushes grafted onto silica nanoparticles via surface reversible addition-fragmentation chain transfer polymerization. Macromolecules 2005, 38, 5929–5936. [Google Scholar]
- Rajesh, R.; Brittain, W.J. Synthesis of high density polymer brushes on nanoparticles by combined RAFT polymerization and click chemistry. Macromol. Rapid Commun. 2008, 29, 1104–1110. [Google Scholar]
- Bindushree, R.; Ranjan, R.; Brittain, W.J. Surface initiated polymerizations from silica nanoparticles. Soft Matter 2006, 2, 386–396. [Google Scholar]
- Von Werne, T.; Patten, T.E. Preparation of structurally well-defined polymer-nanoparticle hybrids with controlled/living radical polymerizations. J. Am. Chem. Soc. 1999, 121, 7409–7410. [Google Scholar] [CrossRef]
- Li, D.J.; Sheng, X.; Zhao, B. Environmentally responsive “hairy” nanoparticles: Mixed homopolymer brushes on silica nanoparticles synthesized by living radical polymerization techniques. J. Am. Chem. Soc. 2005, 127, 6248–6256. [Google Scholar] [CrossRef] [PubMed]
- Perruchot, C.; Khan, M.A.; Kamitsi, A.; Armes, S.V.; von Werne, T.; Patten, T.E. Synthesis of well-defined, polymer-grafted silica particles by aqueous ATRP. Langmuir 2001, 17, 4479–4481. [Google Scholar] [CrossRef]
- Barbey, R.; Lavanant, L.; Paripovic, D.; Schüwer, N.; Sugnaux, C.; Tugulu, S.; Klok, H.-A. Polymer brushes via surface-initiated controlled radical polymerization: Synthesis, characterization, properties, and applications. Chem. Rev. 2009, 109, 5437–5527. [Google Scholar] [CrossRef] [PubMed]
- Ke, M.; Gao, H.; Matyjaszewski, K. Use of ascorbic acid as reducing agent for synthesis of well-defined polymers by ARGET ATRP. Macromolecules 2007, 40, 1789–1791. [Google Scholar]
- Jakubowski, W.; Matyjaszewski, K. Activators regenerated by electron transfer for atom-transfer radical polymerization of (meth)acrylates and related block copolymers. Angew. Chem. Int. Edit. 2006, 45, 4482–4486. [Google Scholar] [CrossRef] [PubMed]
- Abreu, C.M.R.; Mendonca, P.V.; Serra, A.C.; Popov, A.V.; Matyjaszewski, K.; Guliashvili, T.; Coelho, J.F.J. Inorganic sulfites: Efficient reducing agents and supplemental activators for atom transfer radical polymerization. ACS Macro. Lett. 2012, 1, 1308–1311. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Dong, H.C.; Jakubowski, W.; Pietrasik, J.; Kusumo, A. Grafting from surfaces for “everyone”: ARGET ATRP in the presence of air. Langmuir 2007, 23, 4528–4531. [Google Scholar] [CrossRef] [PubMed]
- Hansson, S.; Ostmark, E.; Carlmark, A.; Malmstrom, E. ARGET ATRP for versatile grafting of cellulose using various monomers. ACS Appl. Mater. Inter. 2009, 1, 2651–2659. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, M.; Nystrom, D.; Nordin, O.; Malmstrom, E. Surface modification of thermally expandable microspheres by grafting poly(glycidyl methacrylate) using ARGET ATRP. Eur. Polym. J. 2009, 45, 2374–2382. [Google Scholar] [CrossRef]
- Wang, M.; Yuan, J.; Huang, X.B.; Cai, X.M.; Li, L.; Shen, J. Grafting of carboxybetaine brush onto cellulose membranes via surface-initiated ARGET-ATRP for improving blood compatibility. Colloid Surf. B 2013, 103, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Otsuka, H.; Takahara, A. Poly(methyl methacrylate) grafted imogolite nanotubes prepared through surface-initiated ARGET ATRP. Chem. Commun. 2011, 47, 5813–5815. [Google Scholar] [CrossRef] [PubMed]
- Cheesman, B.T.; Willott, J.D.; Webber, G.B.; Edmondson, S.; Wanless, E.J. Ph-responsive brush-modified silica hybrids synthesized by surface-initiated ARGET ATRP. ACS Macro. Lett. 2012, 1, 1161–1165. [Google Scholar] [CrossRef]
- Zhao, M.N.; Zhou, G.W.; Zhang, L.; Li, X.Y.; Li, T.D.; Liu, F.F. Fabrication and photoactivity of a tunable-void sio2-tio2 core-shell structure on modified sio2 nanospheres by grafting an amphiphilic diblock copolymer using ARGET ATRP. Soft Matter 2014, 10, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Gujadhur, R.; Venkataraman, D.; Kintigh, J.T. Formation of aryl-nitrogen bonds using a soluble copper(I) catalyst. Tetrahedron Lett. 2001, 42, 4791–4793. [Google Scholar] [CrossRef]
- Patel, V.K.; Vishwakarma, N.K.; Mishra, A.K.; Biswas, C.S.; Maiti, P.; Ray, B. Synthesis of alkyne-terminated xanthate raft agents and their uses for the controlled radical polymerization of n-vinylpyrrolidone and the synthesis of its block copolymer using click chemistry. J. Appl. Polym. Sci. 2013, 127, 4305–4317. [Google Scholar] [CrossRef]
- Paoprasert, P.; Spalenka, J.W.; Peterson, D.L.; Ruther, R.E.; Hamers, R.J.; Evans, P.G.; Gopalan, P. Grafting of poly(3-hexylthiophene) brushes on oxides using click chemistry. J. Mater. Chem. 2010, 20, 2651–2658. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Evans, R.A. The rise of azide-alkyne 1,3-dipolar “click” cycloaddition and its application to polymer science and surface modification. Aust. J. Chem. 2007, 60, 384–395. [Google Scholar] [CrossRef]
- Pretsch, C.; Seibl, S. Tables of Spectral Data for Structure Determination of Organic Compounds, 2nd ed.; Sringer-Verlag: Berlin/Heidelberg, Germany, 1989. [Google Scholar]
- Zhuravlev, L.T. The surface chemistry of amorphous silica. Zhuravlev model. Colloids Surf. A 2000, 173, 1–38. [Google Scholar] [CrossRef]
- Tugulu, S.; Barbey, R.; Harms, M.; Fricke, M.; Volkmer, D.; Rossi, A.; Klok, H.A. Synthesis of poly(methacrylic acid) brushes via surface-initiated atom transfer radical polymerization of sodium methacrylate and their use as substrates for the mineralization of calcium carbonate. Macromolecules 2007, 40, 168–177. [Google Scholar] [CrossRef]
- Turgman-Cohen, S.; Genzer, J. Simultaneous bulk- and surface-initiated controlled radical polymerization from planar substrates. J. Am. Chem. Soc. 2011, 133, 17567–17569. [Google Scholar] [CrossRef] [PubMed]
- Gorman, C.B.; Petrie, R.J.; Genzer, J. Effect of substrate geometry on polymer molecular weight and polydispersity during surface-initiated polymerization. Macromolecules 2008, 41, 4856–4865. [Google Scholar] [CrossRef]
- Koylu, D.; Carter, K.R. Stimuli-responsive surfaces utilizing cleavable polymer brush layers. Macromolecules 2009, 42, 8655–8660. [Google Scholar] [CrossRef]
- Devaux, C.; Chapela, J.P.; Beyou, E.; Chaumont, Ph. Controlled structure and density of “living” polystyrene brushes on flat silica surfaces. Eur. Phys. J. 2002, 7, 345–352. [Google Scholar]
- Husseman, M.; Malmström, E.E.; McNamara, M.; Mate, M.; Mecerreyes, D.; Benoit, D.G.; Hedrick, J.L.; Mansky, P.; Huang, E.; Russell, T.P.; et al. Controlled synthesis of polymer brushes by “living” free radical polymerization techniques. Macromolecules 1999, 32, 1424–1431. [Google Scholar] [CrossRef]
- Pasetto, P.; Blas, H.; Audouin, F.; Boissiere, C.; Sanchez, C.; Save, M.; Charleux, B. Mechanistic insight into surface-initiated polymerization of methyl methacrylate and styrene via ATRP from ordered mesoporous silica particles. Macromolecules 2009, 42, 5983–5995. [Google Scholar] [CrossRef]
- Bose, D.S.; Kumar, K.K.; Reddy, A.V.N. A new protocol for selective deprotection of N-tert-butoxycarbonyl protective group (t-Boc) with Sn(OTf)2. Synth. Commun. 2003, 33, 445–450. [Google Scholar] [CrossRef]
- Greene, T.W.; Wuts, P.G.M. Protective Groups in Organic Synthesis, 3rd ed.; John Wiley & Sons Inc.: New York, NY, USA, 1999. [Google Scholar]
- Apelqvist, T.; Wensbo, D. Selective removal of the N-BOC protective group using silica gel at low pressure. Tetrahedron Lett. 1996, 37, 1471–1472. [Google Scholar] [CrossRef]
- Li, B.; Berliner, M.; Buzon, R.; Chiu, C.K.F.; Colgan, S.T.; Kaneko, T.; Keene, N.; Kissel, W.; Le, T.; Leeman, K.R.; et al. Aqueous phosphoric acid as a mild reagent for deprotection of tert-butyl carbamates, esters, and ethers. J. Org. Chem. 2006, 71, 9045–9050. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Bemish, R.; Buzon, R.A.; Chiu, C.K.F.; Colgan, S.T.; Kissel, W.; Le, T.; Leeman, K.R.; Newell, L.; Roth, J. Aqueous phosphoric acid as a mild reagent for deprotection of the t-butoxycarbonyl group. Tetrahedron Lett. 2003, 44, 8113–8115. [Google Scholar] [CrossRef]
- Shin, H.S.; Jung, Y.M.; Chang, T.; Ozaki, Y.; Kim, S.B. Characterization of β-transition of poly(tert-butyl methacrylate) thin films by two-dimensional infrared correlation spectral analysis. Vib. Spectrosc. 2002, 29, 73–77. [Google Scholar] [CrossRef]
- Metaxa, A.F.; Efthimiadou, E.K.; Boukos, N.; Kordas, G. Polysaccharides as a source of advanced materials: Cellulose hollow microspheres for drug delivery in cancer therapy. J. Colloid Interface Sci. 2012, 384, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xu, F.J. Biomolecule-functionalized polymer brushes. Chem. Soc. Rev. 2013, 42, 3394–3426. [Google Scholar] [CrossRef] [PubMed]
- Goddard, J.M.; Hotchkiss, J.H. Polymer surface modification for the attachment of bioactive compounds. Prog. Polym. Sci. 2007, 32, 698–725. [Google Scholar] [CrossRef]
- Ates, Z.; Audouin, F.; Harrington, A.; O’Connor, B.; Heise, A. Functional brush-decorated poly(globalide) films by ARGET-ATRP for bioconjugation. Macromol. Biosci. 2014, 14, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Audouin, F.; Fox, M.; Larragy, R.; Clarke, P.; Huang, J.; O’Connor, B.; Heise, A. Polypeptide-grafted macroporous polyhipe by surface-initiated n-carboxyanhydride (NCA) polymerization as a platform for bioconjugation. Macromolecules 2012, 45, 6127–6135. [Google Scholar] [CrossRef]
- Xu, F.J.; Neoh, K.G.; Kang, E.T. Bioactive surfaces and biomaterials via atom transfer radical polymerization. Prog. Polym. Sci. 2009, 34, 719–761. [Google Scholar] [CrossRef]
- Erickson, H.P. Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online. 2009, 11, 32–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Jia, W.Z.; Qian, Q.Y.; Zhou, Y.G.; Xia, X.H. Simple approach for efficient encapsulation of enzyme in silica matrix with retained bioactivity. Anal. Chem. 2009, 81, 3478–3484. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacono, M.; Heise, A. Stable Poly(methacrylic acid) Brush Decorated Silica Nano-Particles by ARGET ATRP for Bioconjugation. Polymers 2015, 7, 1427-1443. https://doi.org/10.3390/polym7081427
Iacono M, Heise A. Stable Poly(methacrylic acid) Brush Decorated Silica Nano-Particles by ARGET ATRP for Bioconjugation. Polymers. 2015; 7(8):1427-1443. https://doi.org/10.3390/polym7081427
Chicago/Turabian StyleIacono, Marcello, and Andreas Heise. 2015. "Stable Poly(methacrylic acid) Brush Decorated Silica Nano-Particles by ARGET ATRP for Bioconjugation" Polymers 7, no. 8: 1427-1443. https://doi.org/10.3390/polym7081427
APA StyleIacono, M., & Heise, A. (2015). Stable Poly(methacrylic acid) Brush Decorated Silica Nano-Particles by ARGET ATRP for Bioconjugation. Polymers, 7(8), 1427-1443. https://doi.org/10.3390/polym7081427