Flame-Retardancy Properties of Intumescent Ammonium Poly(Phosphate) and Mineral Filler Magnesium Hydroxide in Combination with Graphene


Abstract
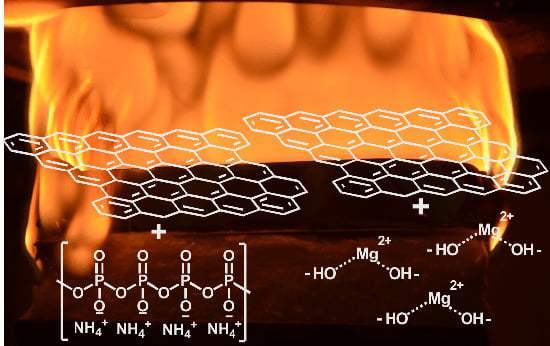
:
1. Introduction
2. Experimental Section
2.1. Materials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | ωPP (wt%) | ωAPP (wt%) | ωMH (wt%) | ωTRGO (wt%) |
|---|---|---|---|---|
| PP | 100 | - | - | - |
| PP/1TRGO | 99 | - | - | 1 |
| PP/APP | 72.5 | 27.5 | - | - |
| PP/APP/0.5TRGO | 72 | 27.5 | - | 0.5 |
| PP/APP/1TRGO | 71.5 | 27.5 | - | 1 |
| PP/APP/2TRGO | 70.5 | 27.5 | - | 2 |
| PP/53MH | 47 | - | 53 | - |
| PP/53MH/1TRGO | 46 | - | 53 | 1 |
| PP/59MH | 41 | - | 59 | - |
| PP/59MH/1TRGO | 40 | - | 59 | 1 |
| PP/60MH | 40 | - | 60 | - |
| PP/60MH/1TRGO | 39 | - | 60 | 1 |
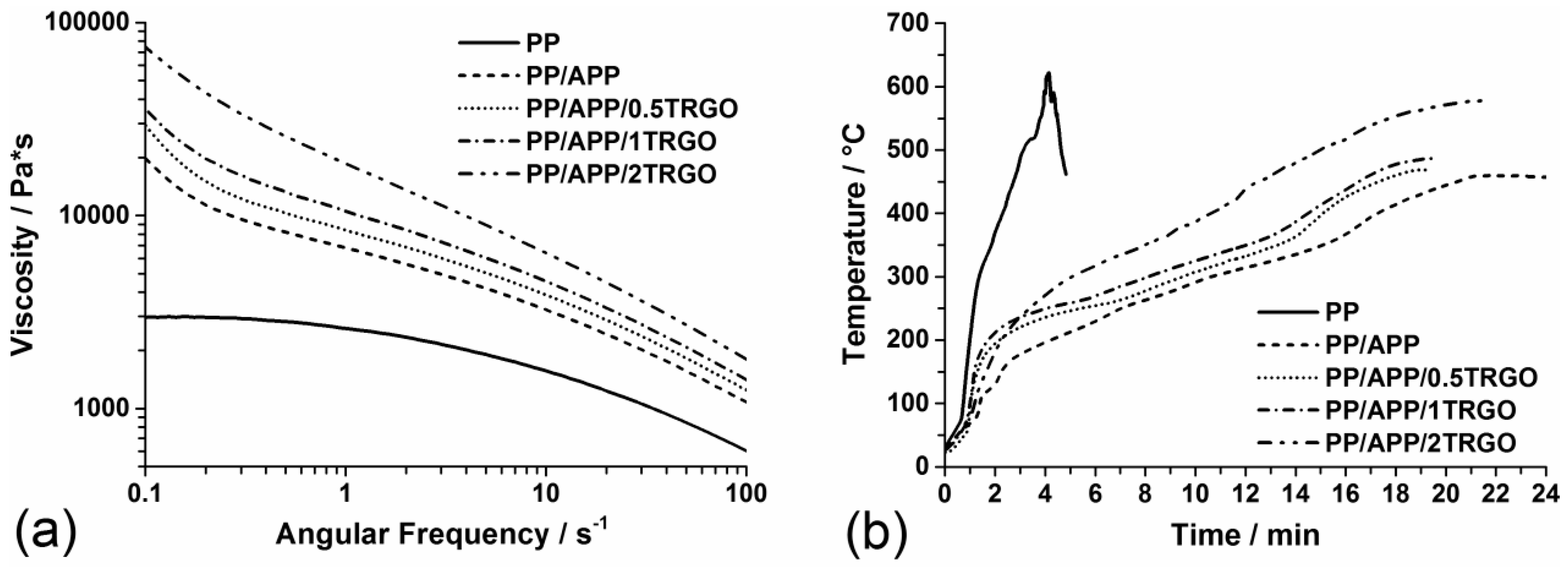
2.2. Methods
2.3. Quantification of Synergism
3. Results and Discussion
3.1. Intumescent APP + TRGO
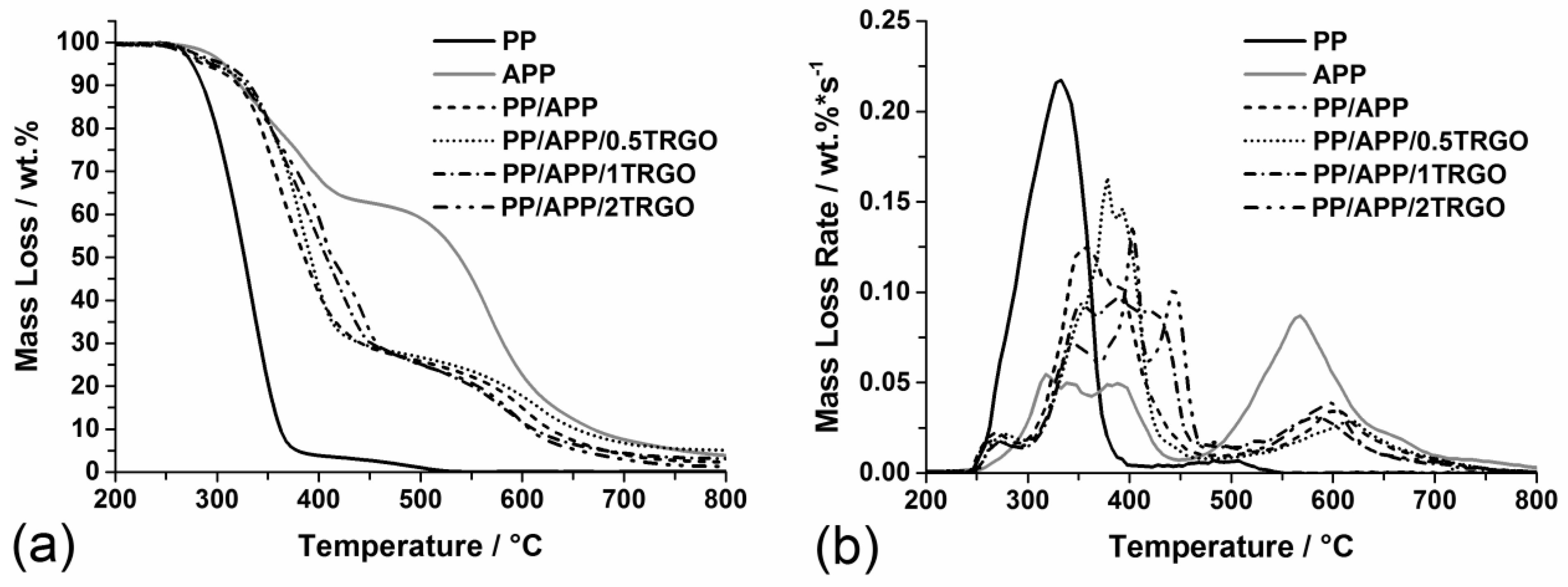
3.1.1. Pyrolysis

3.1.2. Reaction to Small Flame
| Material | OI (vol%) Error ± 1 | UL 94 |
|---|---|---|
| PP | 19 | HB |
| PP/APP | 40 | V-0 |
| PP/APP/0.5TRGO | 36 | V-0 |
| PP/APP/1TRGO | 31 | V-0 |
| PP/APP/2TRGO | 24 | HB |
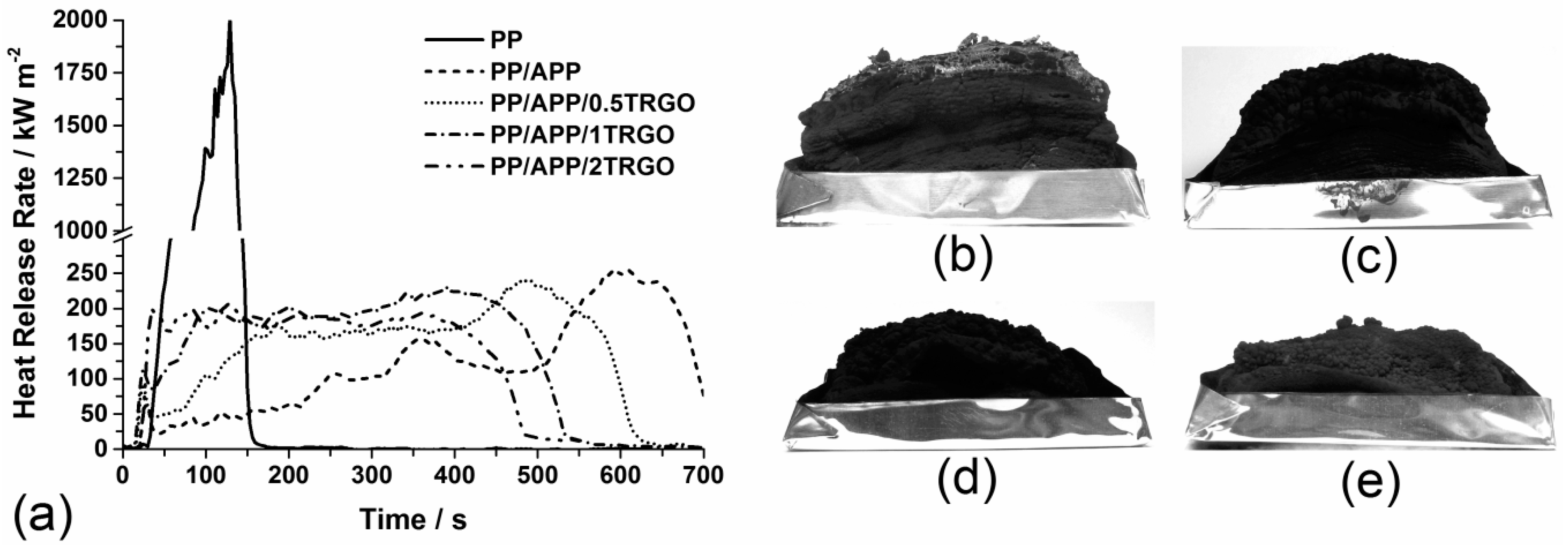
3.1.3. Fire Behavior
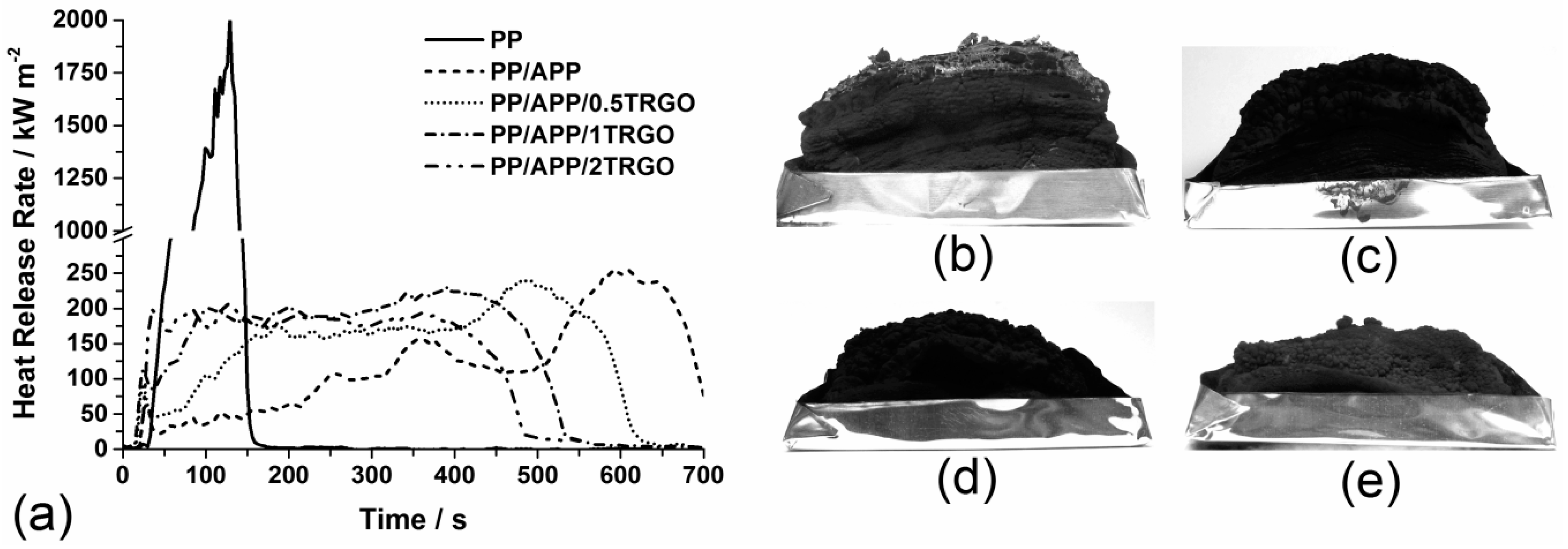
| Material | PHRR (kW/m2) | Residue (wt%) |
|---|---|---|
| PP | 2,011 ± 80 | 0 |
| PP/APP | 254 ± 30 | 14 ± 1 |
| PP/APP/0.5TRGO | 243 ± 10 | 16 ± 1 |
| PP/APP/1TRGO | 230 ± 9 | 16 ± 1 |
| PP/APP/2TRGO | 216 ± 9 | 17 ± 1 |
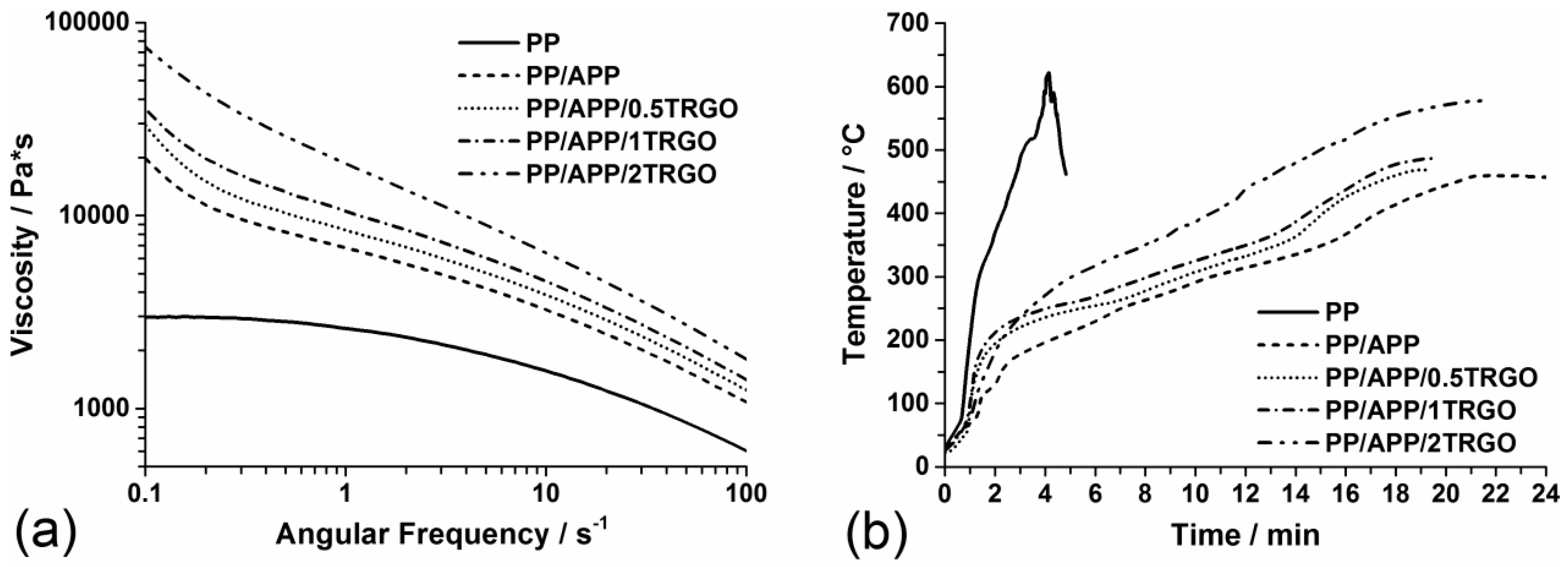
3.1.4. Mechanical Properties
| Material | Young’s modulus (MPa) | Yield stress (MPa) | Charpy notched impact strength (kJ/m²) | SEYoung’s modulus |
|---|---|---|---|---|
| PP | 1,270 ± 15 | 26.8 ± 0.2 | 9.73 ± 0.95 | - |
| PP/APP | 1,840 ± 45 | 20.9 ± 0.4 | 2.36 ± 0.15 | - |
| PP/0.5TRGO | 1,465 ± 32 | 27.6 ± 0.3 | 7.71 ± 0.51 | - |
| PP/1TRGO | 1,465 ± 23 | 27.5 ± 0.1 | 6.90 ± 0.37 | - |
| PP/2TRGO | 1,520 ± 18 | 27.3 ± 0.3 | 5.33 ± 0.15 | - |
| PP/APP/0.5TRGO | 2,050 ± 20 | 22.1 ± 0.1 | 2.64 ± 0.22 | 1.020 |
| PP/APP/1TRGO | 2,080 ± 15 | 22.1 ± 0.1 | 2.37 ± 0.04 | 1.059 |
| PP/APP/2TRGO | 2,005 ± 25 | 22.4 ± 0.2 | 2.09 ± 0.15 | 0.896 |
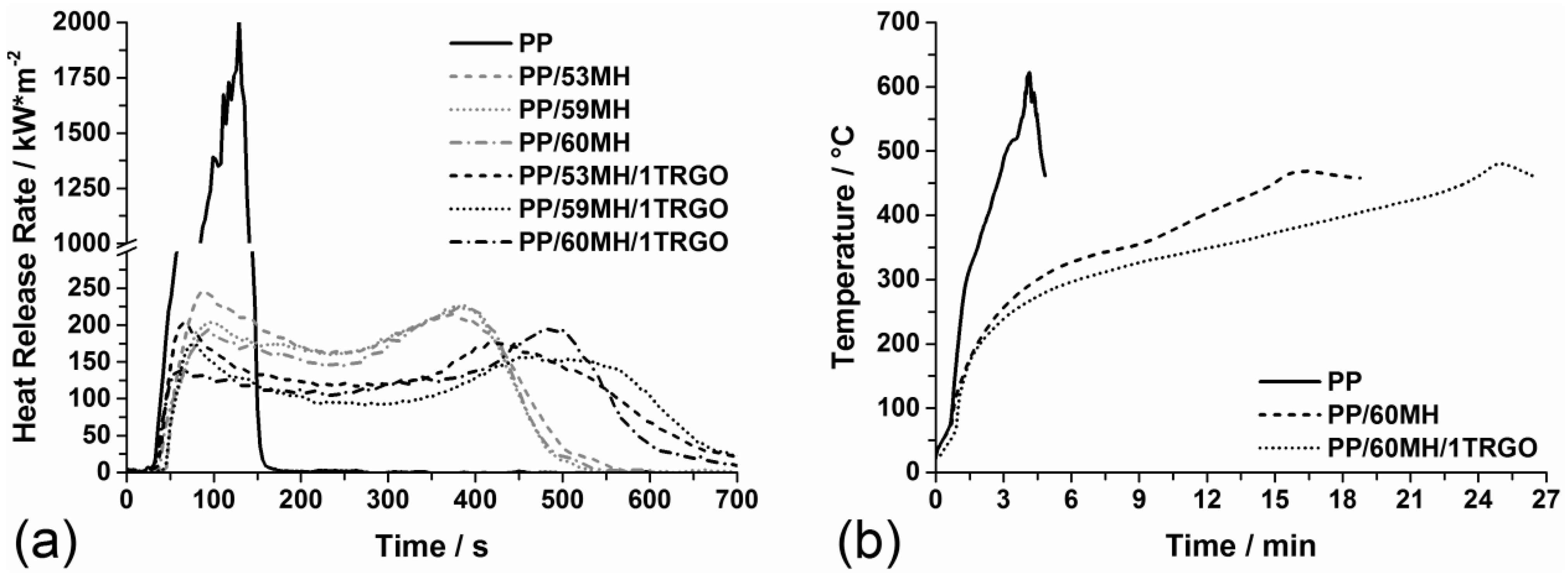
3.2. Mineral Filler Mg(OH)2 + TRGO
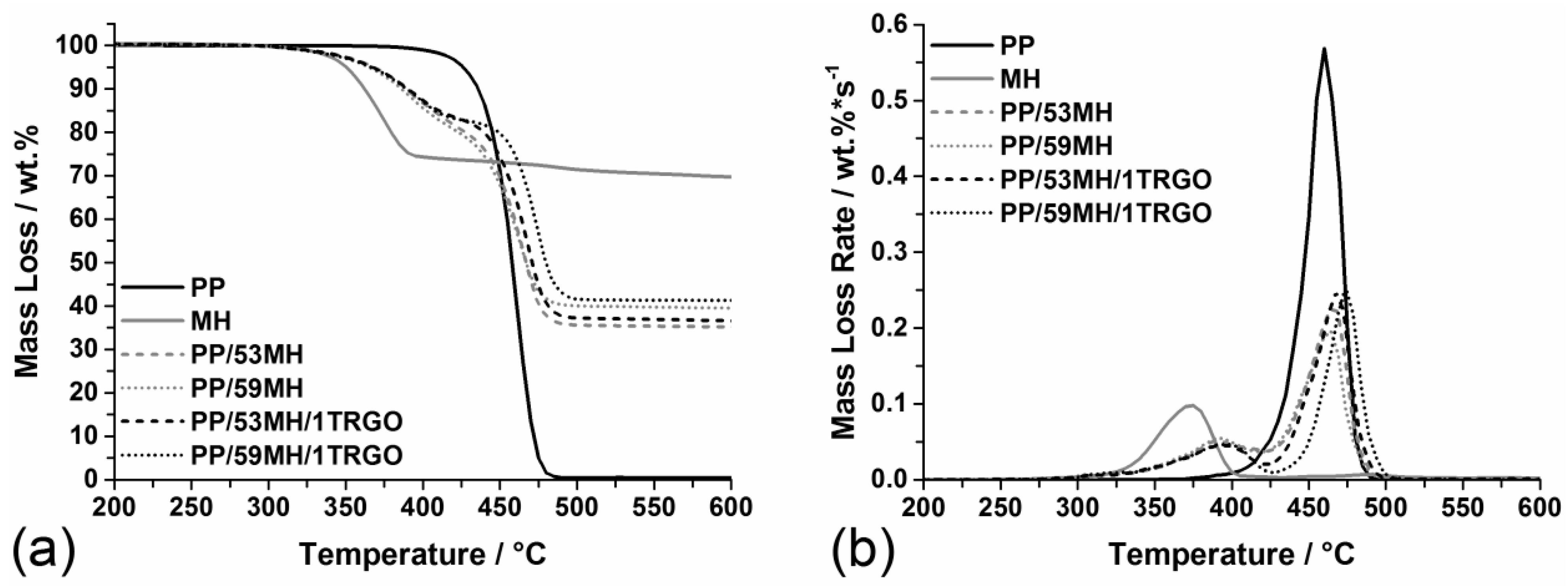
3.2.1. Pyrolysis

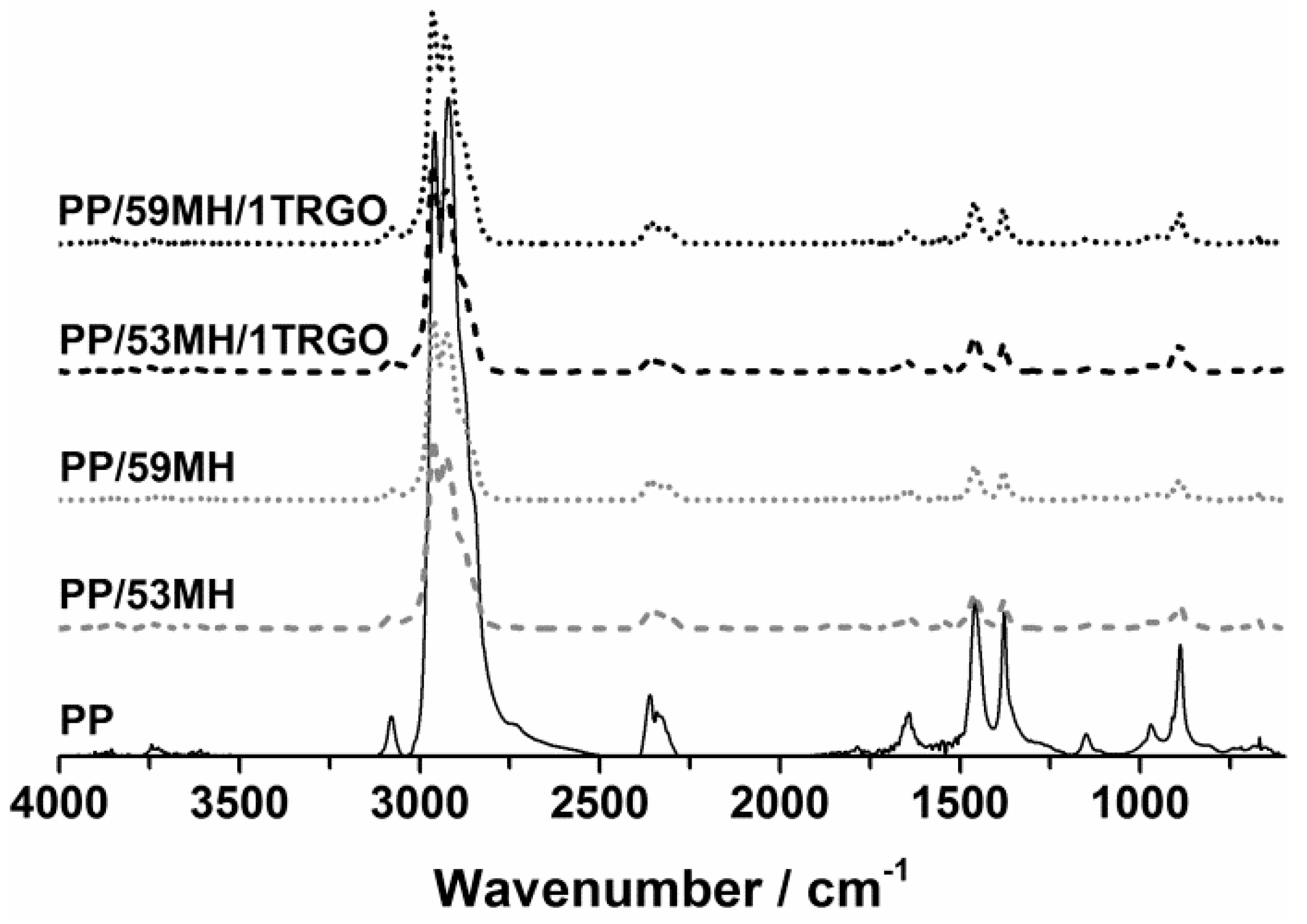
3.2.2. Reaction to Small Flame
| Material | OI (vol%) Error ± 1 | SEOI | UL 94 |
|---|---|---|---|
| PP | 19 | - | HB |
| PP/1TRGO | 19 | - | HB |
| PP/53MH | 28 | - | HB |
| PP/59MH | 32 | - | HB |
| PP/60MH | 31 | - | V-1 |
| PP/53MH/1TRGO | 30 | 1.341 | V-0 |
| PP/59MH/1TRGO | 38 | 1.445 | V-0 |
| PP/60MH/1TRGO | 35 | 1.276 | V-0 |
3.2.3. Fire Behavior
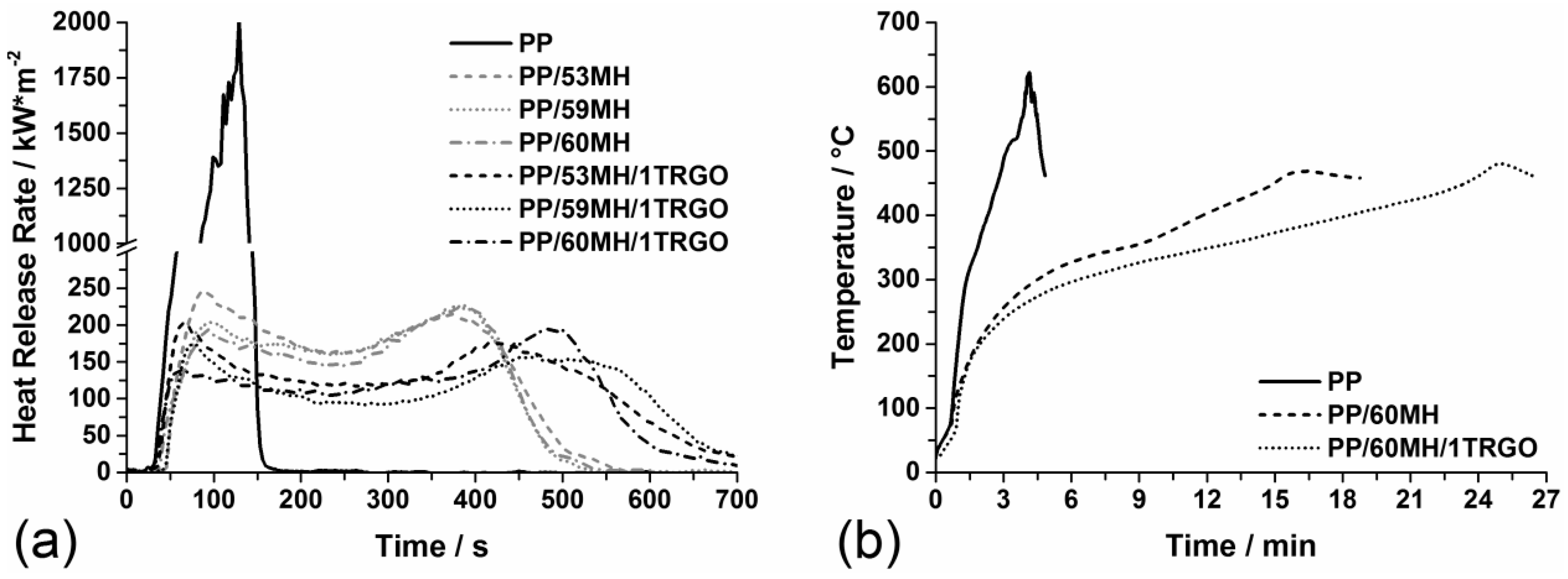
| Material | PHRR (kW/m2) | THE (MJ/m2) | Residue (wt%) | THE/TML (MJ/m2·g) |
|---|---|---|---|---|
| PP | 2,011 ± 80 | 106 ± 4 | 0 | 4.1 ± 0.2 |
| PP/53MH | 240 ± 10 | 81 ± 3 | 33 ± 1 | 3.2 ± 0.1 |
| PP/59MH | 226 ± 9 | 75 ± 3 | 38 ± 2 | 3.0 ± 0.1 |
| PP/60MH | 225 ± 9 | 72 ± 3 | 40 ± 2 | 2.9 ± 0.1 |
| PP/53MH/1G | 203 ± 8 | 82 ± 3 | 34 ± 1 | 3.2 ± 0.1 |
| PP/59MH/1G | 181 ± 7 | 75 ± 3 | 38 ± 2 | 3.0 ± 0.1 |
| PP/60MH/1G | 190 ± 8 | 72 ± 3 | 41 ± 2 | 3.0 ± 0.1 |

3.2.4. Mechanical Properties
| Material | Young’s modulus (MPa) | Yield stress (MPa) | Charpy notched impact strength (kJ/m²) | SEYoung’s modulus |
|---|---|---|---|---|
| PP | 1,270 ± 15 | 26.8 ± 0.2 | 9.73 ± 0.95 | - |
| PP/1TRGO | 1,465 ± 23 | 27.5 ± 0.1 | 6.90 ± 0.37 | - |
| PP/53MH | 2,950 ± 70 | 16.3 ± 0.2 | 29.0 ± 3.10 | - |
| PP/54MH | 2,990 ± 25 | 15.7 ± 0.0 | 35.6 ± 2.62 | - |
| PP/59MH | 3,140 ± 15 | 15.7 ± 0.2 | 29.0 ± 2.46 | - |
| PP/53MH/1G | 3,150 ± 30 | 16.9 ±0.1 | 14.1 ± 0.92 | 1.003 |
| PP/54MH/1G | 3,400 ± 25 | 16.9 ± 0.1 | 13.8 ± 0.95 | 1.113 |
| PP/59MH/1G | 3,500 ± 20 | 16.8 ± 0.0 | 13.4 ± 0.99 | 1.080 |
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Schartel, B. Phosphorus-based flame retardancy mechanisms—Old hat or a starting point for future development? Materials 2010, 3, 4710–4745. [Google Scholar] [CrossRef]
- Levchik, S.V.; Weil, E.D. A review of recent progress in phosphorus-based flame retardants. J. Fire Sci. 2006, 24, 345–364. [Google Scholar] [CrossRef]
- Weil, E.A.; Levchik, S.V. Flame retardants in commercial use or development for polyolefins. J. Fire Sci. 2008, 26, 5–43. [Google Scholar] [CrossRef]
- Bourbigot, S.; Le Bras, M.; Duquesne, S.; Rochery, M. Recent advances for intumescent polymers. Macromol. Mater. Eng. 2004, 289, 499–511. [Google Scholar] [CrossRef]
- Levchik, S.V. Introduction to flame retardancy and polymer flammability. In Flame Retardant Polymer Nanocomposites; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; pp. 1–29. [Google Scholar]
- Camino, G.; Maffezzoli, A.; Braglia, M.; de Lazzaro, M.; Zammarano, M. Effect of hydroxides and hydroxycarbonate structure on fire retardant effectiveness and mechanical properties in ethylene-vinyl acetate copolymer. Polym. Degrad. Stab. 2001, 74, 457–464. [Google Scholar] [CrossRef]
- Hornsby, P.R.; Watson, C.L. Interfacial modification of polypropylene composites filled with magnesium hydroxide. J. Mater. Sci. 1995, 30, 5347–5355. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, F.; Chen, J.; Wang, G.; Liu, H. Investigation of interfacial modification for flame retardant ethylene vinyl acetate copolymer/alumina trihydrate nanocomposites. Polym. Degrad. Stab. 2005, 87, 411–418. [Google Scholar] [CrossRef]
- Hippi, U.; Mattila, J.; Korhonen, M.; Seppälä, J. Compatibilization of polyethylene/aluminum hydroxide (PE/ATH) and polyethylene/magnesium hydroxide (PE/MH) composites with functionalized polyethylenes. Polymer 2003, 44, 1193–1201. [Google Scholar] [CrossRef]
- Gilman, J.W. Flammability and thermal stability studies of polymer layered-silicate (clay) nanocomposites. Appl. Clay Sci. 1999, 15, 31–49. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Grulke, E.; Hilding, J.; Harris, R.; Awad, W.; Douglas, J. Thermal degradation and flammability properties of poly(propylene)/carbon nanotube composites. Macromol. Rapid Commun. 2002, 23, 761–765. [Google Scholar] [CrossRef]
- Gallo, E.; Braun, U.; Schartel, B.; Russo, P.; Acierno, D. Halogen-free flame retarded poly(butylene terephthalate) (PBT) using metal oxides/PBT nanocomposites in combination with aluminium phosphinate. Polym. Degrad. Stab. 2009, 94, 1245–1253. [Google Scholar] [CrossRef]
- Fina, A.; Abbenhuis, H.C.L.; Tabuani, D.; Camino, G. Metal functionalized poss as fire retardants in polypropylene. Polym. Degrad. Stab. 2006, 91, 2275–2281. [Google Scholar] [CrossRef]
- Schartel, B.; Weiss, A. Temperature inside burning polymer specimens: Pyrolysis zone and shielding. Fire Mater. 2010, 34, 217–235. [Google Scholar] [CrossRef]
- Wu, G.M.; Schartel, B.; Bahr, H.; Kleemeier, M.; Yu, D.; Hartwig, A. Experimental and quantitative assessment of flame retardancy by the shielding effect in layered silicate epoxy nanocomposites. Combust. Flame 2012, 159, 3616–3623. [Google Scholar] [CrossRef]
- Dittrich, B.; Wartig, K.A.; Hofmann, D.; Mülhaupt, R.; Schartel, B. Flame retardancy through carbon nanomaterials: Carbon black, multiwall nanotubes, expanded graphite, multi-layer graphene and graphene in polypropylene. Polym. Degrad. Stab. 2013, 98, 1495–1505. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Grulke, E.; Hilding, J.; Groth, K.; Harris, R.; Butler, K.; Shields, J.; Kharchenko, S.; Douglas, J. Thermal and flammability properties of polypropylene/carbon nanotube nanocomposites. Polymer 2004, 45, 4227–4239. [Google Scholar] [CrossRef]
- Schartel, B.; Hull, T.R. Development of fire-retarded materials—Interpretation of cone calorimeter data. Fire Mater. 2007, 31, 327–354. [Google Scholar] [CrossRef]
- Dittrich, B.; Wartig, K.A.; Hofmann, D.; Mülhaupt, R.; Schartel, B. Carbon black, multiwall carbon nanotubes, expanded graphite and functionalized graphene flame retarded polypropylene nanocomposites. Polym. Adv. Technol. 2013, 24, 916–926. [Google Scholar] [CrossRef]
- Dittrich, B.; Wartig, K.A.; Hofmann, D.; Mülhaupt, R.; Schartel, B. The influence of layered, spherical, and tubular carbon nanomaterials’ concentration on the flame retardancy of polypropylene. Polym. Compos. 2014. [Google Scholar] [CrossRef]
- Bartholmai, M.; Schartel, B. Layered silicate polymer nanocomposites: New approach or illusion for fire retardancy? Investigations of the potentials and the tasks using a model system. Polym. Adv. Technol. 2004, 15, 355–364. [Google Scholar] [CrossRef]
- Schartel, B.; Pötschke, P.; Knoll, U.; Abdel-Goad, M. Fire behaviour of polyamide 6/multiwall carbon nanotube nanocomposites. Eur. Polym. J. 2005, 41, 1061–1070. [Google Scholar] [CrossRef]
- Schartel, B.; Braun, U.; Knoll, U.; Bartholmai, M.; Goering, H.; Neubert, D.; Potschke, P. Mechanical, thermal, and fire behavior of bisphenol a polycarbonate/multiwall carbon nanotube nanocomposites. Polym. Eng. Sci. 2008, 48, 149–158. [Google Scholar] [CrossRef]
- Bourbigot, S.; Bras, M.L.; Dabrowski, F.; Gilman, J.W.; Kashiwagi, T. PA-6 clay nanocomposite hybrid as char forming agent in intumescent formulations. Fire Mater. 2000, 24, 201–208. [Google Scholar] [CrossRef]
- Pawlowski, K.H.; Schartel, B. Flame retardancy mechanisms of aryl phosphates in combination with boehmite in bisphenol a polycarbonate/acrylonitrile-butadiene-styrene blends. Polym. Degrad. Stab. 2008, 93, 657–667. [Google Scholar] [CrossRef]
- Vannier, A.; Duquesne, S.; Bourbigot, S.; Castrovinci, A.; Camino, G.; Delobel, R. The use of POSS as synergist in intumescent recycled poly(ethylene terephthalate). Polym. Degrad. Stab. 2008, 93, 818–826. [Google Scholar] [CrossRef]
- Beyer, G. Flame retardancy of nanocomposites based on organoclays and carbon nanotubes with aluminium trihydrate. Polym. Adv. Technol. 2006, 17, 218–225. [Google Scholar] [CrossRef]
- Schartel, B.; Knoll, U.; Hartwig, A.; Pütz, D. Phosphonium-modified layered silicate epoxy resins nanocomposites and their combinations with ATH and organo-phosphorus fire retardants. Polym. Adv. Technol. 2006, 17, 281–293. [Google Scholar] [CrossRef]
- Gallo, E.; Schartel, B.; Acierno, D.; Russo, P. Flame retardant biocomposites: Synergism between phosphinate and nanometric metal oxides. Eur. Polym. J. 2011, 47, 1390–1401. [Google Scholar] [CrossRef]
- Lu, H.D.; Wilkie, C.A. Study on intumescent flame retarded polystyrene composites with improved flame retardancy. Polym. Degrad. Stab. 2010, 95, 2388–2395. [Google Scholar] [CrossRef]
- Hummers, W.S.; Offeman, R.E. Preparation of graphitic oxide. J. Am. Chem. Soc. 1958, 80, 1339. [Google Scholar] [CrossRef]
- Tölle, F.J.; Fabritius, M.; Mülhaupt, R. Emulsifier-free graphene dispersions with high graphene content for printed electronics and freestanding graphene films. Adv. Funct. Mater. 2012, 22, 1136–1144. [Google Scholar] [CrossRef]
- Steurer, P.; Wissert, R.; Thomann, R.; Mülhaupt, R. Functionalized graphenes and thermoplastic nanocomposites based upon expanded graphite oxide. Macromol. Rapid Commun. 2009, 30, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Kalaitzidou, K.; Fukushima, H.; Drzal, L.T. A new compounding method for exfoliated graphite-polypropylene nanocomposites with enhanced flexural properties and lower percolation threshold. Compos. Sci. Technol. 2007, 67, 2045–2051. [Google Scholar] [CrossRef]
- Fina, A.; Cuttica, F.; Camino, G. Ignition of polypropylene/montmorillonite nanocomposites. Polym. Degrad. Stab. 2012, 97, 2619–2626. [Google Scholar] [CrossRef]
- Weil, E.D. Synergists, adjuvants and antagonists in flame-retardant systems. In Fire Retardancy of Polymeric Materials; CRC Press: Boca Raton, FL, USA, 2000; pp. 115–145. [Google Scholar]
- Despinasse, M.C.; Schartel, B. Aryl phosphate–aryl phosphate synergy in flame-retarded bisphenol a polycarbonate/acrylonitrile-butadiene-styrene. Thermochim. Acta 2013, 563, 51–61. [Google Scholar] [CrossRef]
- Horrocks, A.R.; Smart, G.; Nazaré, S.; Kandola, B.; Price, D. Quantification of zinc hydroxystannate and stannate synergies in halogen-containing flame-retardant polymeric formulations. J. Fire Sci. 2009. [Google Scholar] [CrossRef]
- Wu, G.M.; Schartel, B.; Yu, D.; Kleemeier, M.; Hartwig, A. Synergistic fire retardancy in layered-silicate nanocomposite combined with low-melting phenysiloxane glass. J. Fire Sci. 2012, 30, 69–87. [Google Scholar] [CrossRef]
- Zanetti, M.; Bracco, P.; Costa, L. Thermal degradation behaviour of PE/clay nanocomposites. Polym. Degrad. Stab. 2004, 85, 657–665. [Google Scholar] [CrossRef]
- Levchik, S.; Wilkie, C.A. Char formation. In Fire Retardancy of Polymeric Materials; CRC Press: Boca Raton, FL, USA, 2000; pp. 171–215. [Google Scholar]
- Adams, J.H. Analysis of the nonvolatile oxidation products of polypropylene I. Thermal oxidation. J. Polym. Sci. A Polym. Chem. 1970, 8, 1077–1090. [Google Scholar] [CrossRef]
- François-Heude, A.; Richaud, E.; Leprovost, J.; Heninger, M.; Mestdagh, H.; Desnoux, E.; Colin, X. Real-time quantitative analysis of volatile products generated during solid-state polypropylene thermal oxidation. Polym. Test. 2013, 32, 907–917. [Google Scholar] [CrossRef]
- Hoff, A.; Jacobsson, S. Thermal oxidation of polypropylene in the temperature range of 120–280 °C. J. Appl. Polym. Sci. 1984, 29, 465–480. [Google Scholar] [CrossRef]
- Camino, G.; Costa, L.; Trossarelli, L. Study of the mechanism of intumescence in fire retardant polymers: Part I—Thermal degradation of ammonium polyphosphate-pentaerythritol mixtures. Polym. Degrad. Stab. 1984, 6, 243–252. [Google Scholar] [CrossRef]
- Duquesne, S.; Le Bras, M.; Bourbigot, S.; Delobel, R.; Camino, G.; Eling, B.; Lindsay, C.; Roels, T.; Vezin, H. Mechanism of fire retardancy of polyurethanes using ammonium polyphosphate. J. Appl. Polym. Sci. 2001, 82, 3262–3274. [Google Scholar] [CrossRef]
- Schartel, B.; Weiß, A.; Mohr, F.; Kleemeier, M.; Hartwig, A.; Braun, U. Flame retarded epoxy resins by adding layered silicate in combination with the conventional protection-layer-building flame retardants melamine borate and ammonium polyphosphate. J. Appl. Polym. Sci. 2010, 118, 1134–1143. [Google Scholar]
- Bugajny, M.; Bras, M.L.; Bourbigot, S. New approach to the dynamic properties of an intumescent material. Fire Mater. 1999, 23, 49–51. [Google Scholar] [CrossRef]
- Jimenez, M.; Duquesne, S.; Bourbigot, S. High-throughput fire testing for intumescent coatings. Ind. Eng. Chem. Res. 2006, 45, 7475–7481. [Google Scholar] [CrossRef]
- Duquesne, S.; Magnet, S.; Jama, C.; Delobel, R. Intumescent paints: Fire protective coatings for metallic substrates. Surf. Coat. Technol. 2004, 180–181, 302–307. [Google Scholar] [CrossRef]
- Bartholmai, M.; Schriever, R.; Schartel, B. Influence of external heat flux and coating thickness on the thermal insulation properties of two different intumescent coatings using cone calorimeter and numerical analysis. Fire Mater. 2003, 27, 151–162. [Google Scholar] [CrossRef]
- Bartholmai, M.; Schartel, B. Assessing the performance of intumescent coatings using bench-scaled cone calorimeter and finite difference simulations. Fire Mater. 2007, 31, 187–205. [Google Scholar] [CrossRef]
- Hornsby, P.R.; Mthupha, A. Rheological characterization of polypropylene filled with magnesium hydroxide. J. Mater. Sci. 1994, 29, 5293–5301. [Google Scholar] [CrossRef]
- Kim, S. Flame retardancy and smoke suppression of magnesium hydroxide filled polyethylene. J. Polym. Sci. B Polym. Phys. 2003, 41, 936–944. [Google Scholar] [CrossRef]
- Haurie, L.; Fernández, A.I.; Velasco, J.I.; Chimenos, J.M.; Lopez Cuesta, J.M.; Espiell, F. Thermal stability and flame retardancy of LDPE/EVA blends filled with synthetic hydromagnesite/aluminium hydroxide/montmorillonite and magnesium hydroxide/aluminium hydroxide/montmorillonite mixtures. Polym. Degrad. Stabil. 2007, 92, 1082–1087. [Google Scholar] [CrossRef]
- Braun, U.; Schartel, B. Flame retardant mechanisms of red phosphorus and magnesium hydroxide in high impact polystyrene. Macromol. Chem. Phys. 2004, 205, 2185–2196. [Google Scholar] [CrossRef]
- Beyer, G. Flame retardant properties of eva-nanocomposites and improvements by combination of nanofillers with aluminium trihydrate. Fire Mater. 2001, 25, 193–197. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Sumi, K. Thermal decomposition products of polypropylene. J. Polym. Sci. 1969, 7, 1599–1607. [Google Scholar] [CrossRef]
- Kiran, E.; Gillham, J.K. Pyrolysis-molecular weight chromatography: A new on-line system for analysis of polymers. II. Thermal decomposition of polyolefins: Polyethylene, polypropylene, polyisobutylene. J. Appl. Polym. Sci. 1976, 20, 2045–2068. [Google Scholar] [CrossRef]
- Hornsby, P.R. The application of magnesium hydroxide as a fire retardant and smoke-suppressing additive for polymers. Fire Mater. 1994, 18, 269–276. [Google Scholar] [CrossRef]
- Rothon, R.N.; Hornsby, P.R. Flame retardant effects of magnesium hydroxide. Polym. Degrad. Stab. 1996, 54, 383–385. [Google Scholar] [CrossRef]
- Wu, G.M.; Schartel, B.; Kleemeier, M.; Hartwig, A. Flammability of layered silicate epoxy nanocomposites combined with low-melting inorganic ceepree glass. Polym. Eng. Sci. 2012, 52, 507–517. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dittrich, B.; Wartig, K.-A.; Mülhaupt, R.; Schartel, B. Flame-Retardancy Properties of Intumescent Ammonium Poly(Phosphate) and Mineral Filler Magnesium Hydroxide in Combination with Graphene. Polymers 2014, 6, 2875-2895. https://doi.org/10.3390/polym6112875
Dittrich B, Wartig K-A, Mülhaupt R, Schartel B. Flame-Retardancy Properties of Intumescent Ammonium Poly(Phosphate) and Mineral Filler Magnesium Hydroxide in Combination with Graphene. Polymers. 2014; 6(11):2875-2895. https://doi.org/10.3390/polym6112875
Chicago/Turabian StyleDittrich, Bettina, Karen-Alessa Wartig, Rolf Mülhaupt, and Bernhard Schartel. 2014. "Flame-Retardancy Properties of Intumescent Ammonium Poly(Phosphate) and Mineral Filler Magnesium Hydroxide in Combination with Graphene" Polymers 6, no. 11: 2875-2895. https://doi.org/10.3390/polym6112875
APA StyleDittrich, B., Wartig, K.-A., Mülhaupt, R., & Schartel, B. (2014). Flame-Retardancy Properties of Intumescent Ammonium Poly(Phosphate) and Mineral Filler Magnesium Hydroxide in Combination with Graphene. Polymers, 6(11), 2875-2895. https://doi.org/10.3390/polym6112875