Coarse-Grained Models for Protein-Cell Membrane Interactions

Abstract
:1. Introduction
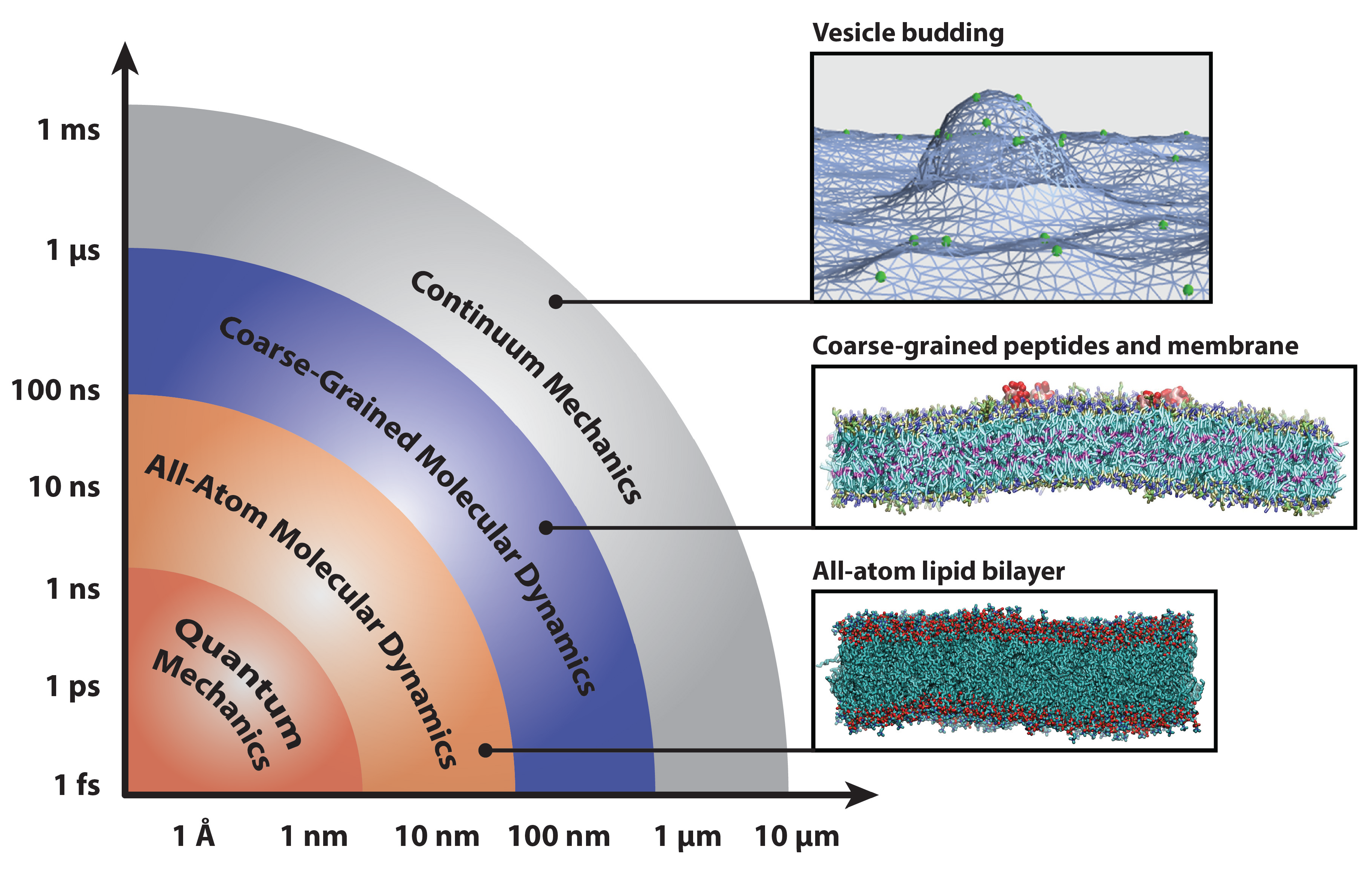
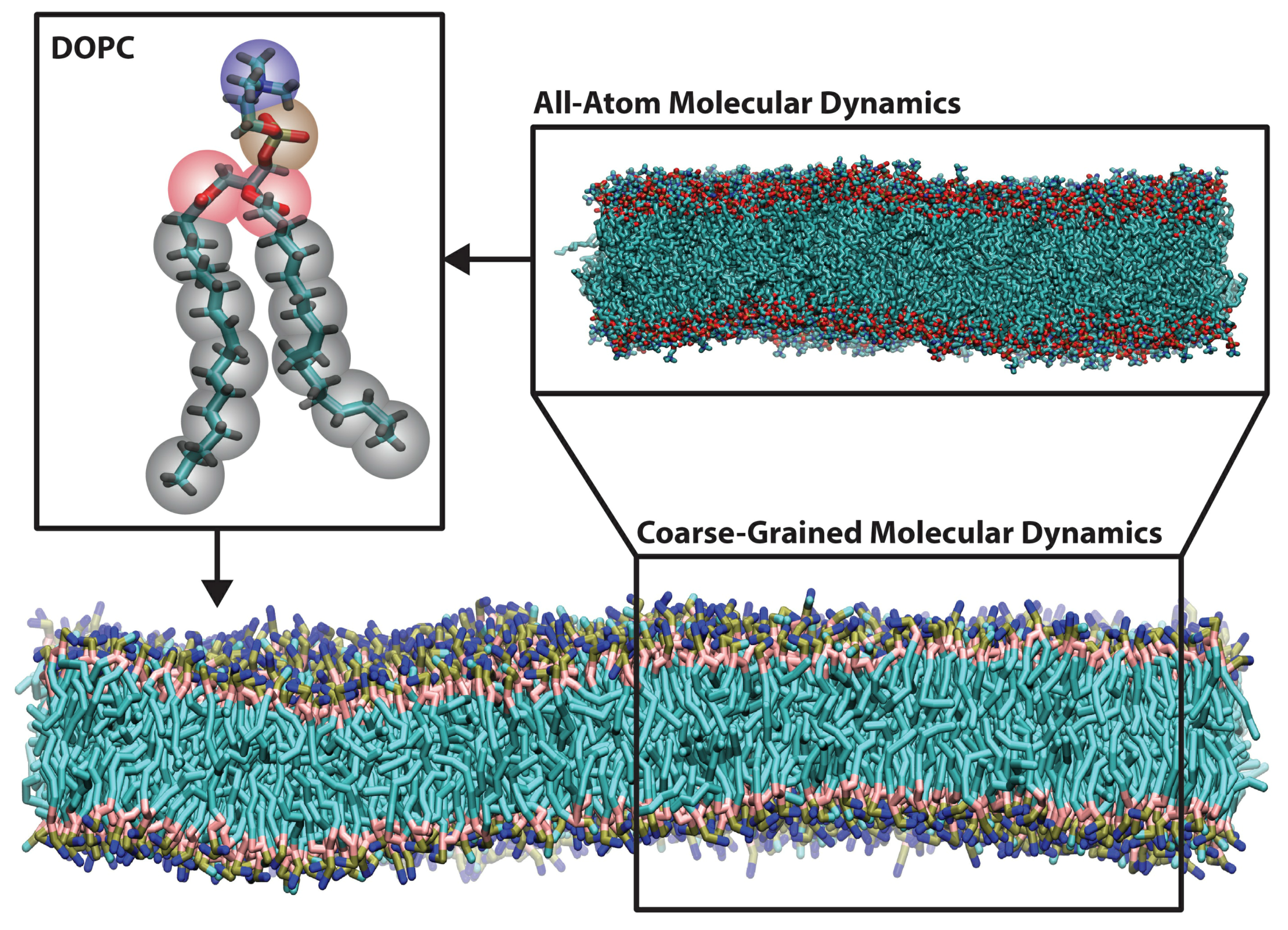
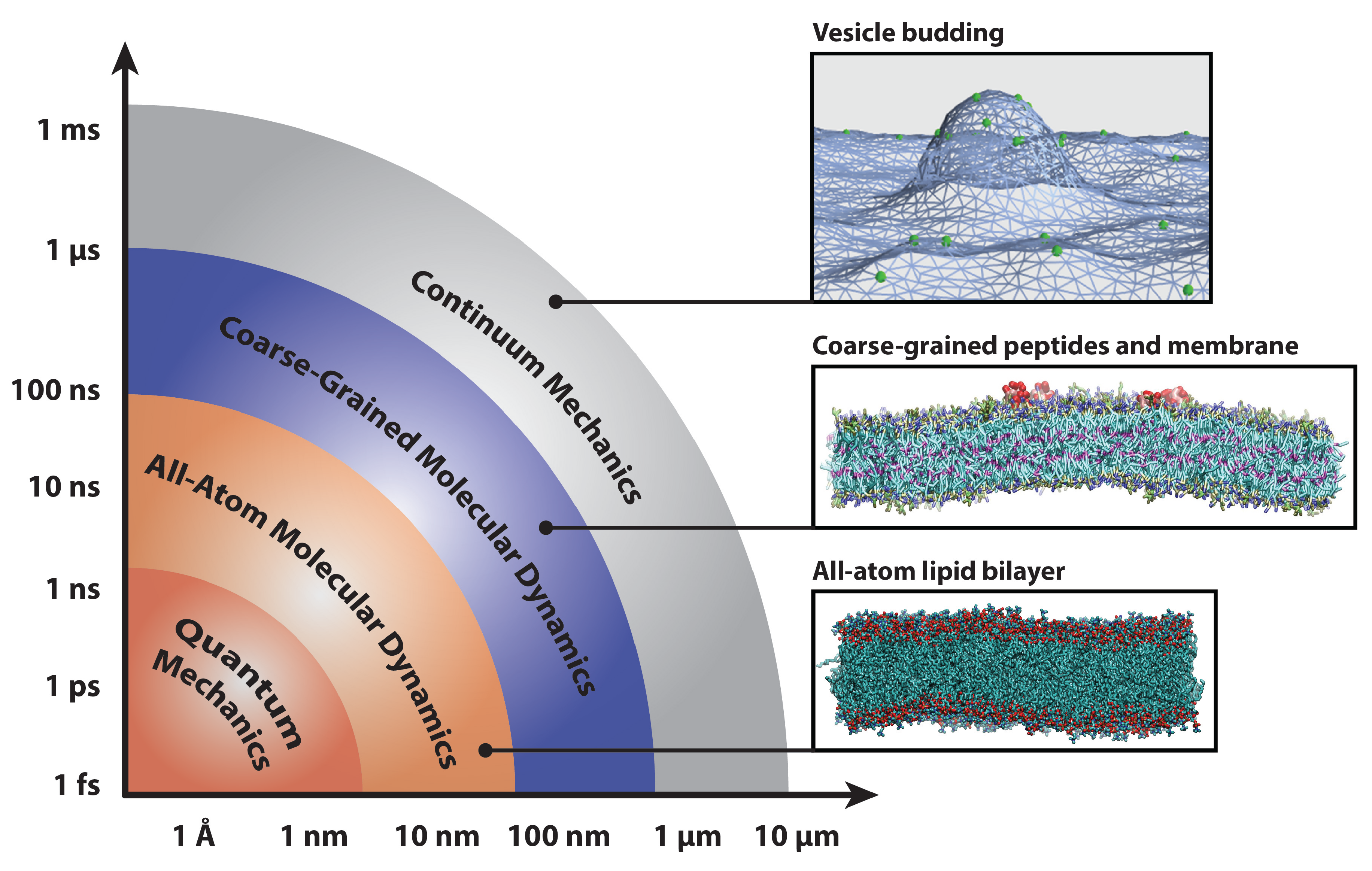
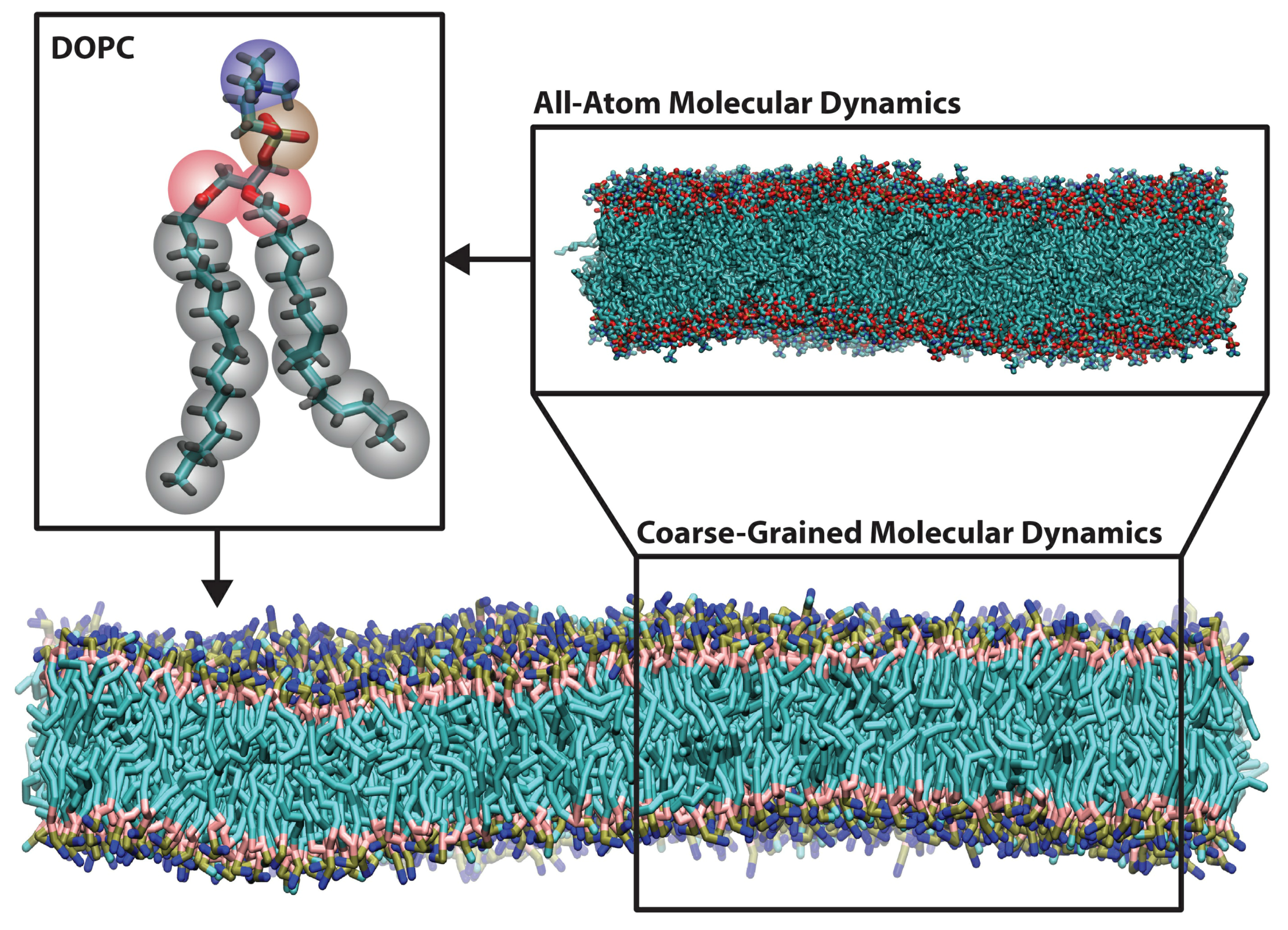
2. Methods for Parameterizing Coarse-Grained Force Fields
2.1. Early Coarse-Grained Models and Dissipative Particle Dynamics
2.2. Structure and Energy Matching in the CMM-CG Model
2.3. Force Matching with the Multiscale Coarse Grained (MS-CG) Model
2.4. The Energy-Based Approach of the Martini Force Field
2.5. Reproducing Experiments in Coarse-Grained Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Key Methods | Key Target Data |
|---|---|---|
| CMM-CG [44,69] | structure matching, energy matching, Boltzmann inversion, reverse Monte Carlo | density distributions, interfacial tension, area per lipid, bending modulus, area compressibility modulus, lipid order parameters |
| MS-CG [49,55] | bottom-up force matching, variational optimization, cubic spline basis functions, hybrid analytic-systematic coarse-graining, screened electrostatics | atomistic site-to-site radial distribution functions, density distributions, bending modulus, area compressibility modulus, lipid diffusion rates |
| Martini [16,60] | top-down energy matching, potential of mean force between phases, bilayer stress profile, free energy of lipid desorption or flip-flop, short-range electrostatics | free energy of hydration, free energy of vaporization, partitioning free energies, surface tension, interfacial tension, density distributions, bending modulus, area per lipid |
2.6. Assessing CGMD Model Performance
| Property | Experimental Method | Simulation Measurement |
|---|---|---|
| partition coefficient | titration calorimetry | potential of mean force of a particle pulled between phases |
| self-diffusion coefficient | magnetic resonance spin echo | mean-squared displacement |
| electron density profile | X-ray scattering | electron density |
| area per lipid | neutron scattering | area measurement (bilayer mid-plane) |
| lipid order parameter | nuclear magnetic resonance (NMR) | lipid tail angles to the bilayer normal |
| phase transition temperature | cryo-transmission electron microscopy (cryo-TEM) | structure factor |
| pressure-area isotherm | Langmuir trough, captive bubble surfactometer | pressure tensor, area measurement |
| line tension | fluorescence microscopy of GUVs, micropipette aspiration | pressure tensor |
| bending rigidity | video phase contrast microscopy, GUV shear flow | height-height fluctuation spectrum |
3. Modeling Proteins
3.1. Atomistic Simulations of Proteins
3.1.1. Enhanced Sampling Methods
3.1.2. Atomistic Simulations of Membrane Proteins
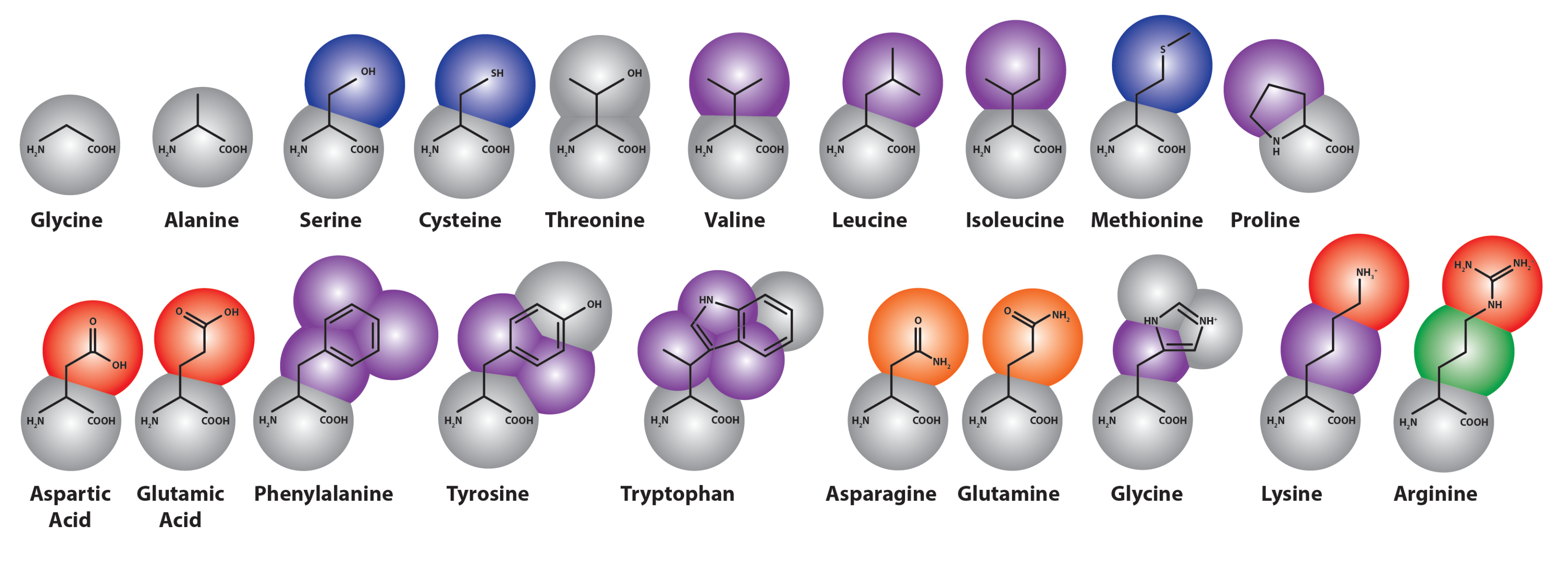
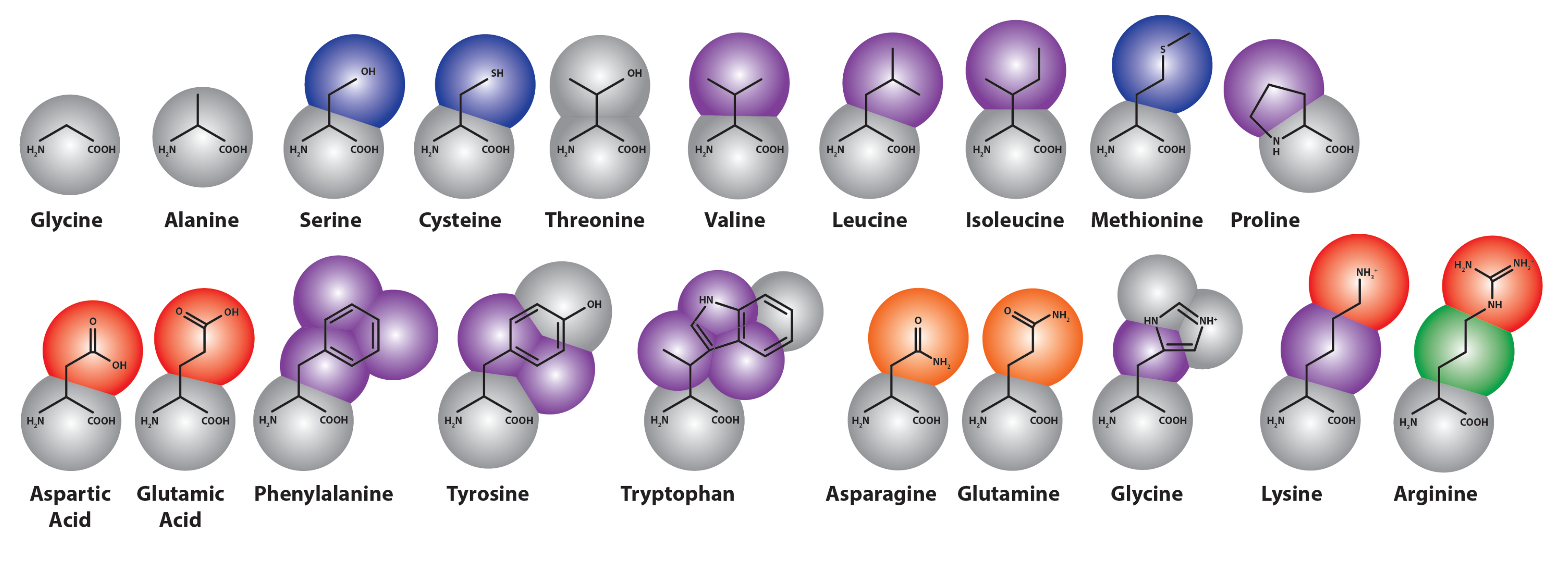
3.2. Parameterization of Coarse-Grained Proteins
3.2.1. Structure-Based Coarse-Grained Protein Modeling
3.2.2. Martini Proteins
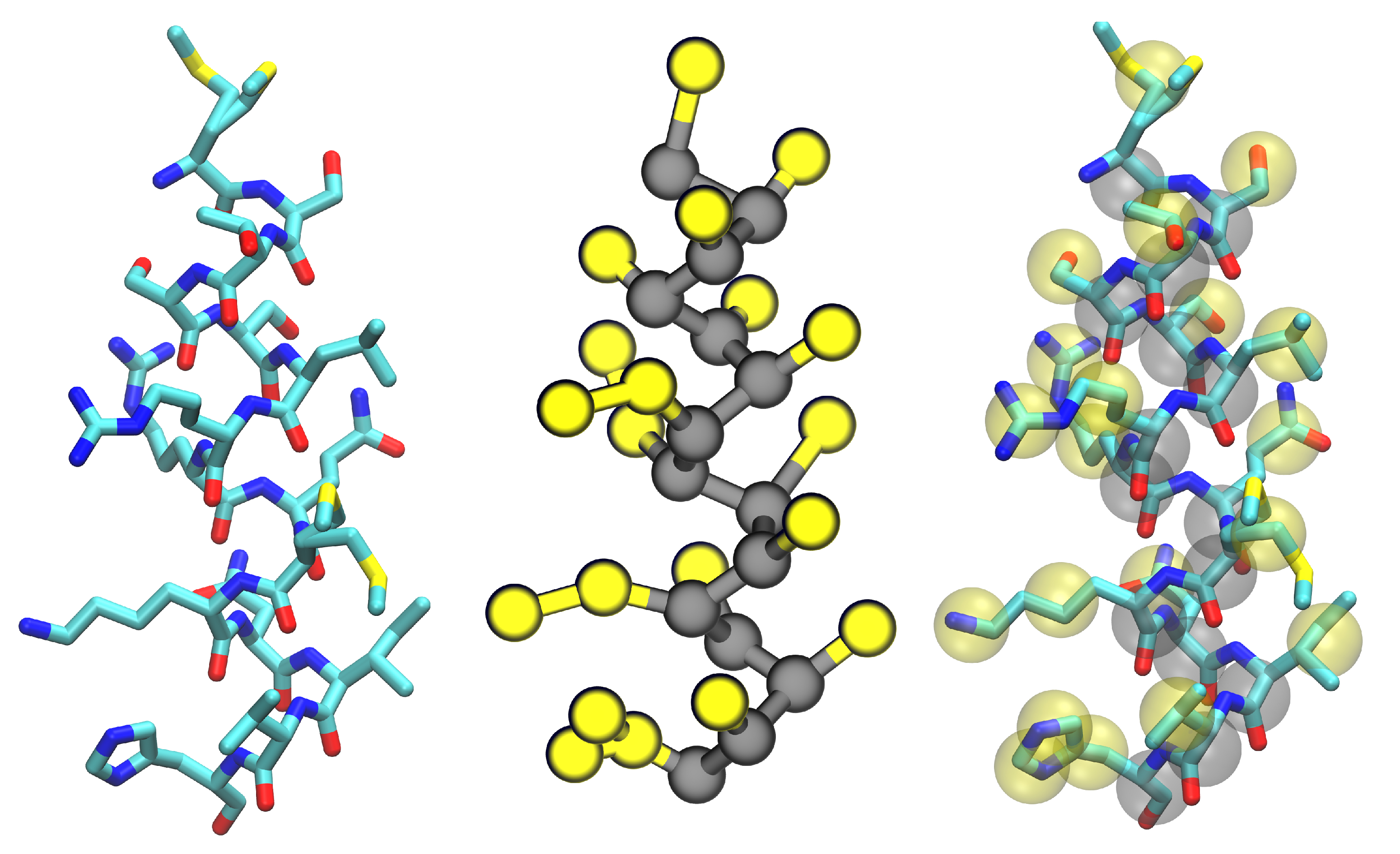
3.3. Improvements to Protein Models
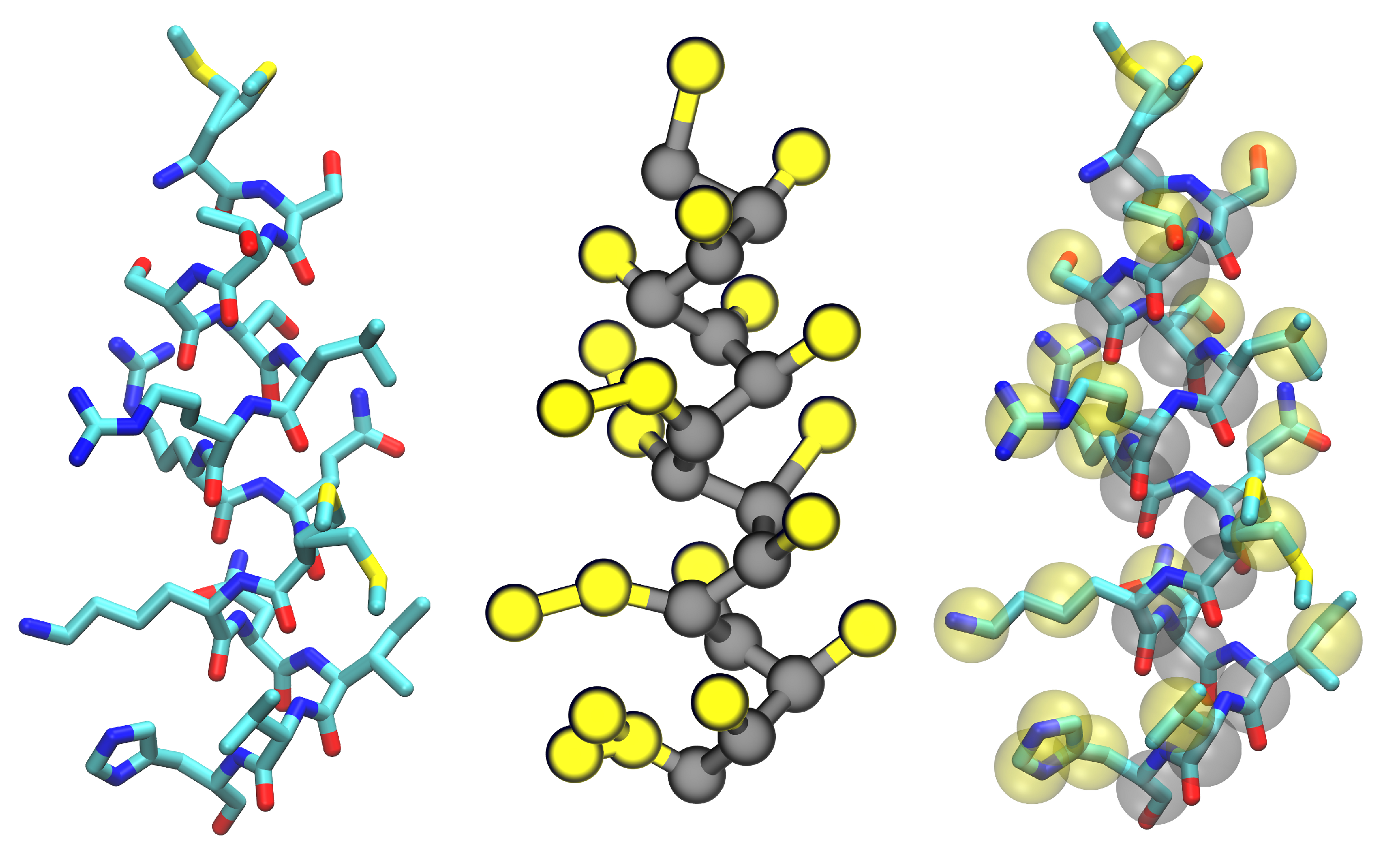
4. Membrane-Protein Applications
4.1. Simulations of Biological Membranes
4.2. Modeling Membrane Bending
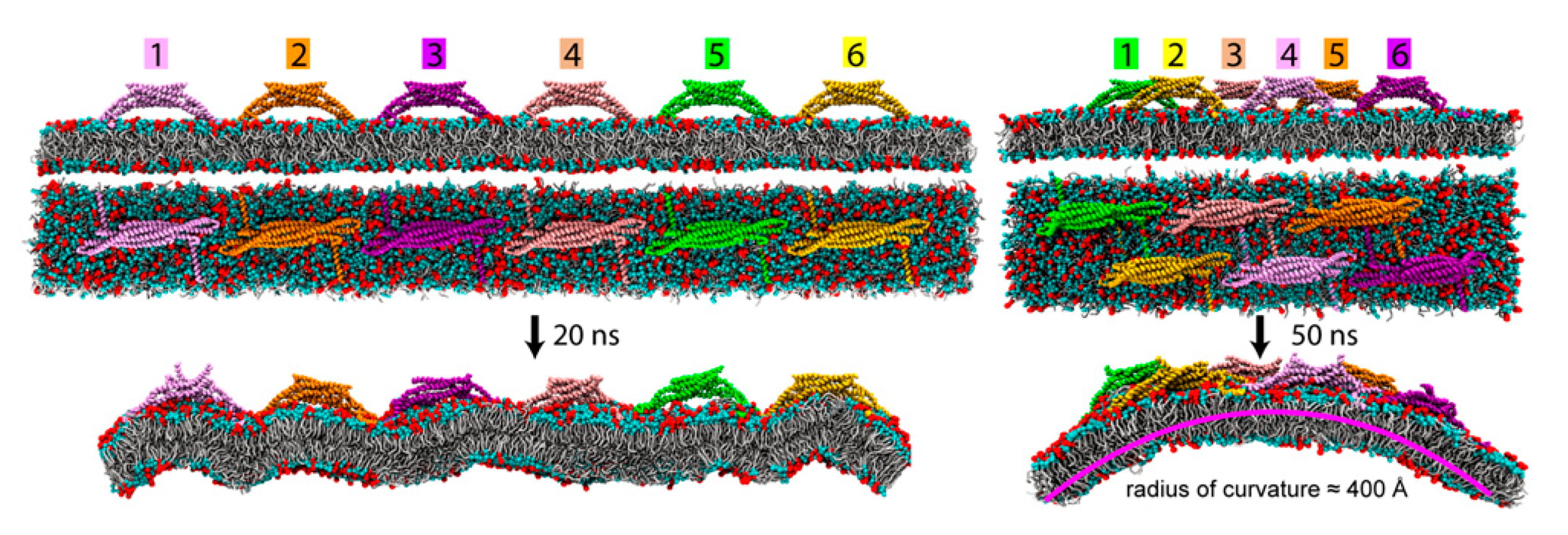
4.3. Lipid Bilayers Support Protein Assembly and Function
4.4. Extending to the Mesoscale
5. Conclusions and Future Directions
Acknowledgments
Conflict of Interest
References
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Arkhipov, A.; Yin, Y.; Schulten, K. Membrane-bending mechanism of amphiphysin N-BAR domains. Biophys. J. 2009, 97, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Yefimov, S.; van der Giessen, E.; Onck, P.R.; Marrink, S.J. Mechanosensitive membrane channels in action. Biophys. J. 2008, 94, 2994–3002. [Google Scholar] [CrossRef] [PubMed]
- a Nijeholt, J.A.L.; Bulacu, M.; Marrink, S.J.; Driessen, A.J.M. Immobilization of the plug domain inside the SecY channel allows unrestricted protein translocation. J. Biol. Chem. 2010, 285, 23747–23754. [Google Scholar] [CrossRef] [PubMed]
- Bond, P.J.; Sansom, M.S.P. Bilayer deformation by the Kv channel voltage sensor domain revealed by self-assembly simulations. Proc. Natl. Acad. Sci. USA 2007, 104, 2631–2636. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Freddolino, P.L.; Arkhipov, A.; Schulten, K. Assembly of lipoprotein particles revealed by coarse-grained molecular dynamics simulations. J. Struct. Biol. 2007, 157, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Khurana, E.; DeVane, R.H.; Kohlmeyer, A.; Klein, M.L. Probing peptide nanotube self-assembly at a liquid-liquid interface with coarse-grained molecular dynamics. Nano Lett. 2008, 8, 3626–3630. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Marrink, S.J. Lipid-mediated interactions tune the association of glycophorin A helix and its disruptive mutants in membranes. Phys. Chem. Chem. Phys. 2010, 12, 12987–12996. [Google Scholar] [CrossRef] [PubMed]
- Ulmschneider, J.P.; Andersson, M.; Ulmschneider, M.B. Determining peptide partitioning properties via computer simulation. J. Membr. Biol. 2011, 239, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmans, M.; Knecht, V.; Marrink, S.J. A single bicontinuous cubic phase induced by fusion peptides. J. Am. Chem. Soc. 2009, 131, 9166–9167. [Google Scholar] [CrossRef] [PubMed]
- Baoukina, S.; Tieleman, D.P. Direct simulation of protein-mediated vesicle fusion: Lung surfactant protein B. Biophys. J. 2010, 99, 2134–2142. [Google Scholar] [CrossRef] [PubMed]
- Shillcock, J. Vesicles and Vesicle Fusion: Coarse-Grained Simulations. In Methods in Molecular Biology; Monticelli, L., Salonen, E., Eds.; Humana Press: New York, NY, USA, 2013; Volume 924, pp. 659–697. [Google Scholar]
- Psachoulia, E.; Fowler, P.W.; Bond, P.J.; Sansom, M.S.P. Helix-helix interactions in membrane proteins: Coarse-grained simulations of glycophorin a helix dimerization. Biochemistry 2008, 47, 10503–10512. [Google Scholar] [CrossRef] [PubMed]
- Balabin, I.A.; Yang, W.; Beratan, D.N. Coarse-grained modeling of allosteric regulation in protein receptors. Proc. Natl. Acad. Sci. USA 2009, 106, 14253–14258. [Google Scholar] [CrossRef] [PubMed]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Klepeis, J.L.; Lindorff-Larsen, K.; Dror, R.O.; Shaw, D.E. Long-timescale molecular dynamics simulations of protein structure and function. Curr. Opin. Struct. Biol. 2009, 19, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, M.S.; Eastman, P.; Vaidyanathan, V.; Houston, M.; Legrand, S.; Beberg, A.L.; Ensign, D.L.; Bruns, C.M.; Pande, V.S. Accelerating molecular dynamic simulation on graphics processing units. J. Comput. Chem. 2009, 30, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Freddolino, P.L.; Harrison, C.B.; Liu, Y.; Schulten, K. Challenges in protein-folding simulations. Nat. Phys. 2010, 6, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Izvekov, S.; Chung, P.W.; Rice, B.M. The multiscale coarse-graining method: Assessing its accuracy and introducing density dependent coarse-grain potentials. J. Chem. Phys. 2010, 133, 064109:1–064109:16. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, I.F.; Goldenberg, D.P.; Voth, G.A. Exploration of transferability in multiscale coarse-grained peptide models. J. Phys. Chem. B 2011, 115, 11911–11926. [Google Scholar] [CrossRef] [PubMed]
- Engin, O.; Villa, A.; Peter, C.; Sayar, M. A challenge for peptide coarse graining: Transferability of fragment-based models. Macromol. Theory Simul. 2011, 20, 451–465. [Google Scholar] [CrossRef]
- Li, Y.; Abberton, B.C.; Kroger, M.; Liu, W.K. Challenges in multiscale modeling of polymer dynamics. Polymers 2013, 5, 751–832. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Müller, M.; Katsov, K.; Schick, M. Coarse-grained models and collective phenomena in membranes: Computer simulation of membrane fusion. J. Polym. Sci. B Polym. Phys. 2003, 41, 1441–1450. [Google Scholar] [CrossRef]
- Shelley, J.C.; Shelley, M.Y. Computer simulation of surfactant solutions. Curr. Opin. Colloid Interface Sci. 2000, 5, 101–110. [Google Scholar] [CrossRef]
- Larson, R. Monte Carlo lattice simulation of amphiphilic systems in two and three dimensions. J. Chem. Phys. 1988, 89, 1642–1650. [Google Scholar] [CrossRef]
- Smit, B.; Hilbers, P.A.J.; Esselink, K.; Rupert, L.A.M.; van Os, N.M.; Schlijper, A.G. Computer simulations of a water/oil interface in the presence of micelles. Nature 1990, 348, 624–625. [Google Scholar] [CrossRef]
- Goetz, R.; Lipowsky, R. Computer simulations of bilayer membranes: Self-assembly and interfacial tension. J. Chem. Phys. 1998, 108, 7397–7409. [Google Scholar] [CrossRef]
- Goetz, R.; Gompper, G.; Lipowsky, R. Mobility and elasticity of self-assembled membranes. Phys. Rev. Lett. 1999, 82, 221–224. [Google Scholar] [CrossRef]
- Hoogerbrugge, P.J.; Koelman, J.M.V.A. Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics. Europhys. Lett. 1992, 19. [Google Scholar] [CrossRef]
- Español, P.; Warren, P. Statistical mechanics of dissipative particle dynamics. Europhys. Lett. 1995, 30. [Google Scholar] [CrossRef]
- Groot, R.D.; Warren, P.B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Venturoli, M.; Smit, B. Simulating the self-assembly of model membranes. PhysChemComm 1999, 2, 45–49. [Google Scholar] [CrossRef]
- Grafmüller, A.; Shillcock, J.; Lipowsky, R. Pathway of membrane fusion with two tension-dependent energy barriers. Phys. Rev. Lett. 2007, 98, 218101:1–218101:4. [Google Scholar] [CrossRef]
- Venturoli, M. Simulation studies of protein-induced bilayer deformations, and lipid-induced protein tilting, on a mesoscopic model for lipid bilayers with embedded proteins. Biophys. J. 2005, 88, 1778–1798. [Google Scholar] [CrossRef] [PubMed]
- Groot, R.; Rabone, K. Mesoscopic simulation of cell membrane damage, morphology change and rupture by nonionic surfactants. Biophys. J. 2001, 81, 725–736. [Google Scholar] [CrossRef]
- Venturoli, M.; Maddalena Sperotto, M.; Kranenburg, M.; Smit, B. Mesoscopic models of biological membranes. Phys. Rep. 2006, 437, 1–54. [Google Scholar] [CrossRef]
- Goga, N.; Rzepiela, A.J.; de Vries, A.H.; Marrink, S.J.; Berendsen, H.J.C. Efficient algorithms for langevin and DPD dynamics. J. Chem. Theory Comput. 2012, 8, 3637–3649. [Google Scholar] [CrossRef]
- Shelley, J.C.; Shelley, M.Y.; Reeder, R.C.; Bandyopadhyay, S.; Klein, M.L. A coarse grain model for phospholipid simulations. J. Phys. Chem. B 2001, 105, 4464–4470. [Google Scholar] [CrossRef]
- Feller, S.E.; MacKerell, A.D. An improved empirical potential energy function for molecular simulations of phospholipids. J. Phys. Chem. B 2000, 104, 7510–7515. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; van Buuren, A.; Apol, E.; Meulenhoff, P.; Tieleman, D.; Sijbers, A.; Feenstra, K.; van Drunen, R.; et al. Gromacs User Manual version 4.0. Available online: http://www.gromacs.org/ (accessed on 28 June 2013).
- Lyubartsev, A.P.; Laaksonen, A. Calculation of effective interaction potentials from radial distribution functions: A reverse Monte Carlo approach. Phys. Rev. E 1995, 52, 3730–3737. [Google Scholar] [CrossRef]
- Demond, A.H.; Lindner, A.S. Estimation of interfacial tension between organic liquids and water. Environ. Sci. Technol. 1993, 27, 2318–2331. [Google Scholar] [CrossRef]
- Izvekov, S.; Voth, G.A. A multiscale coarse-graining method for biomolecular systems. J. Phys. Chem. B 2005, 109, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Izvekov, S.; Parrinello, M.; Burnham, C.J.; Voth, G.A. Effective force fields for condensed phase systems from ab initio molecular dynamics simulation: A new method for force-matching. J. Chem. Phys. 2004, 120, 10896:1–10896:18. [Google Scholar] [CrossRef] [PubMed]
- Izvekov, S.; Violi, A.; Voth, G.A. Systematic coarse-graining of nanoparticle interactions in molecular dynamics simulation. J. Phys. Chem. B 2005, 109, 17019–17024. [Google Scholar] [CrossRef] [PubMed]
- Ercolessi, F.; Adams, J.B. Interatomic potentials from first-principles calculations: The force-matching method. Europhys. Lett. 1994, 26, 583. [Google Scholar] [CrossRef]
- Beers, K.J. Numerical Methods for Chemical Engineering: Applications in Matlab; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Orsi, M.; Haubertin, D.Y.; Sanderson, W.E.; Essex, J.W. A quantitative coarse-grain model for lipid bilayers. J. Phys. Chem. B 2008, 112, 802–815. [Google Scholar] [CrossRef] [PubMed]
- Ayton, G.S.; Voth, G.A. Hybrid coarse-graining approach for lipid bilayers at large length and time scales. J. Phys. Chem. B 2009, 113, 4413–4424. [Google Scholar] [CrossRef] [PubMed]
- Ayton, G.S.; Lyman, E.; Voth, G.A. Hierarchical coarse-graining strategy for protein-membrane systems to access mesoscopic scales. Faraday Discuss. 2010, 144, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Copestake, A.P.; Neilson, G.W.; Enderby, J.E. The structure of a highly concentrated aqueous solution of lithium chloride. J. Phys. C Solid State Phys. 1985, 18. [Google Scholar] [CrossRef]
- Lide, D. CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 1992. [Google Scholar]
- Christen, M.; Hünenberger, P.H.; Bakowies, D.; Baron, R.; Bürgi, R.; Geerke, D.P.; Heinz, T.N.; Kastenholz, M.A.; Kräutler, V.; Oostenbrink, C.; et al. The GROMOS software for biomolecular simulation: GROMOS05. J. Comput. Chem. 2005, 26, 1719–1751. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; de Vries, A.H.; Mark, A.E. Coarse grained model for semiquantitative lipid simulations. J. Phys. Chem. B 2004, 108, 750–760. [Google Scholar] [CrossRef]
- Schatzberg, P. Solubilities of water in several normal alkanes from C7 to C161. J. Phys. Chem. 1963, 67, 776–779. [Google Scholar] [CrossRef]
- Viswanadhan, V.N.; Ghose, A.K.; Singh, U.C.; Wendoloski, J.J. Prediction of solvation free energies of small organic molecules additive-constitutive models based on molecular fingerprints and atomic constants. J. Chem. Inf. Comput. Sci. 1999, 39, 405–412. [Google Scholar] [CrossRef]
- Duffy, E.M.; Jorgensen, W.L. Prediction of properties from simulations: Free energies of solvation in hexadecane, octanol, and water. J. Am. Chem. Soc. 2000, 122, 2878–2888. [Google Scholar] [CrossRef]
- Douglass, D.C.; McCall, D.W. Diffusion in paraffin hydrocarbons. J. Phys. Chem. 1958, 62, 1102–1107. [Google Scholar] [CrossRef]
- Krynicki, K.; Green, C.D.; Sawyer, D.W. Pressure and temperature dependence of self-diffusion in water. Faraday Discuss. Chem. Soc. 1978, 66, 199–208. [Google Scholar] [CrossRef]
- Baron, R.; Trzesniak, D.; de Vries, A.H.; Elsener, A.; Marrink, S.J.; van Gunsteren, W.F. Comparison of thermodynamic properties of coarse-grained and atomic-level simulation models. ChemPhysChem 2007, 8, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Amaya, J.; Rana, D.; Hornof, V. Dynamic interfacial tension behavior of water/oil systems containing in situ-formed surfactants. J. Solut. Chem. 2002, 31, 139–148. [Google Scholar] [CrossRef]
- Nagle, J.F.; Tristram-Nagle, S. Structure of lipid bilayers. Biochim. Biophys. Acta Biomembr. 2000, 1469, 159–195. [Google Scholar] [CrossRef]
- Shinoda, W.; DeVane, R.; Klein, M.L. Zwitterionic lipid assemblies: Molecular dynamics studies of monolayers, bilayers, and vesicles using a new coarse grain force field. J. Phys. Chem. B 2010, 114, 6836–6849. [Google Scholar] [CrossRef] [PubMed]
- Ben-Naim, A. Solvation Thermodynamics; Plenum Press: New York, NY, USA, 1987. [Google Scholar]
- Rowe, E.S.; Zhang, F.; Leung, T.W.; Parr, J.S.; Guy, P.T. Thermodynamics of membrane partitioning for a series of n-alcohols determined by titration calorimetry: Role of hydrophobic effects. Biochemistry 1998, 37, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Ulmschneider, M.B.; Doux, J.P.F.; Killian, J.A.; Smith, J.C.; Ulmschneider, J.P. Mechanism and kinetics of peptide partitioning into membranes from all-atom simulations of thermostable peptides. J. Am. Chem. Soc. 2010, 132, 3452–3460. [Google Scholar] [CrossRef] [PubMed]
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar]
- Yesylevskyy, S.; Schäfer, L.; Sengupta, D.; Marrink, S. Polarizable water model for the coarse-grained MARTINI force field. PLoS Comput. Biol. 2010, 6, e1000810. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.E.; Barnes, A.C.; Salmon, P.S. Neutron and X-ray diffraction studies of liquids and glasses. Rep. Prog. Phys. 2006, 69, 233. [Google Scholar] [CrossRef]
- Kučerka, N.; Nagle, J.F.; Sachs, J.N.; Feller, S.E.; Pencer, J.; Jackson, A.; Katsaras, J. Lipid bilayer structure determined by the simultaneous analysis of neutron and X-ray scattering data. Biophys. J. 2008, 95, 2356–2367. [Google Scholar] [CrossRef] [PubMed]
- Mills, T.T.; Tristram-Nagle, S.; Heberle, F.A.; Morales, N.F.; Zhao, J.; Wu, J.; Toombes, G.E.; Nagle, J.F.; Feigenson, G.W. Liquid-liquid domains in bilayers detected by wide angle X-ray scattering. Biophys. J. 2008, 95, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, J.D.; Sachs, J.N. Experimental verification of lipid bilayer structure through multi-scale modeling. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 2284–2290. [Google Scholar] [CrossRef] [PubMed]
- Piggot, T.J.; Piñeiro, Á.; Khalid, S. Molecular dynamics simulations of phosphatidylcholine membranes: A comparative force field study. J. Chem. Theory Comput. 2012, 8, 4593–4609. [Google Scholar] [CrossRef]
- Seelig, A.; Seelig, J. Dynamic structure of fatty acyl chains in a phospholipid bilayer measured by deuterium magnetic resonance. Biochemistry 1974, 13, 4839–4845. [Google Scholar] [CrossRef] [PubMed]
- McConnell, H.M. Structures and transitions in lipid monolayers at the air-water interface. Annu. Rev. Phys. Chem. 1991, 42, 171–195. [Google Scholar] [CrossRef]
- Siegel, D.; Epand, R. The mechanism of lamellar-to-inverted hexagonal phase transitions in phosphatidylethanolamine: Implications for membrane fusion mechanisms. Biophys. J. 1997, 73, 3089–3111. [Google Scholar] [CrossRef]
- Kharakoz, D.P.; Shlyapnikova, E.A. Thermodynamics and kinetics of the early steps of solid-state nucleation in the fluid lipid bilayer. J. Phys. Chem. B 2000, 104, 10368–10378. [Google Scholar]
- Baoukina, S.; Monticelli, L.; Marrink, S.J.; Tieleman, D.P. Pressure-area isotherm of a lipid monolayer from molecular dynamics simulations. Langmuir 2007, 23, 12617–12623. [Google Scholar] [CrossRef] [PubMed]
- Baoukina, S.; Monticelli, L.; Risselada, H.J.; Marrink, S.J.; Tieleman, D.P. The molecular mechanism of lipid monolayer collapse. Proc. Natl. Acad. Sci. USA 2008, 105, 10803–10808. [Google Scholar] [PubMed]
- Baumgart, T.; Hess, S.T.; Webb, W.W. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature 2003, 425, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Tian, A.; Johnson, C.; Wang, W.; Baumgart, T. Line tension at fluid membrane domain boundaries measured by micropipette aspiration. Phys. Rev. Lett. 2007, 98, 208102:1–208102:4. [Google Scholar] [CrossRef]
- Sonne, J.; Hansen, F.Y.; Peters, G.H. Methodological problems in pressure profile calculations for lipid bilayers. J. Chem. Phys. 2005, 122, 124903:1–124903:9. [Google Scholar] [CrossRef] [PubMed]
- Hamai, C.; Yang, T.; Kataoka, S.; Cremer, P.S.; Musser, S.M. Effect of average phospholipid curvature on supported bilayer formation on glass by vesicle fusion. Biophys. J. 2006, 90, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Ollila, O.H.S.; Risselada, H.J.; Louhivuori, M.; Lindahl, E.; Vattulainen, I.; Marrink, S.J. 3D pressure field in lipid membranes and membrane-protein complexes. Phys. Rev. Lett. 2009, 102, 078101:1–078101:4. [Google Scholar] [CrossRef]
- Lindahl, E.; Edholm, O. Spatial and energetic-entropic decomposition of surface tension in lipid bilayers from molecular dynamics simulations. J. Chem. Phys. 2000, 113, 3882–3893. [Google Scholar] [CrossRef]
- Jähnig, F. What is the surface tension of a lipid bilayer membrane? Biophys. J. 1996, 71, 1348–1349. [Google Scholar] [CrossRef]
- Feller, S.E.; Pastor, R.W. Constant surface tension simulations of lipid bilayers: The sensitivity of surface areas and compressibilities. J. Chem. Phys. 1999, 111, 1281–1287. [Google Scholar] [CrossRef]
- Marrink, S.J.; Mark, A.E. Effect of undulations on surface tension in simulated bilayers. J. Phys. Chem. B 2001, 105, 6122–6127. [Google Scholar] [CrossRef]
- Sonne, J.; Jensen, M.Ø.; Hansen, F.Y.; Hemmingsen, L.; Peters, G.H. Reparameterization of all-atom dipalmitoylphosphatidylcholine lipid parameters enables simulation of fluid bilayers at zero tension. Biophys. J. 2007, 92, 4157–4167. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, W. Elastic properties of lipid bilayers: Theory and possible experiments. Z. Naturforsch. C Biosci. 1973, 28, 693–703. [Google Scholar]
- Niggemann, G.; Kummrow, M.; Helfrich, W. The bending rigidity of phosphatidylcholine bilayers: Dependences on experimental method, sample cell sealing and temperature. J. Phys. II Fr. 1995, 5, 413–425. [Google Scholar] [CrossRef]
- De Haas, K.H.; Blom, C.; van den Ende, D.; Duits, M.H.G.; Mellema, J. Deformation of giant lipid bilayer vesicles in shear flow. Phys. Rev. E 1997, 56, 7132–7137. [Google Scholar] [CrossRef]
- Rawicz, W.; Olbrich, K.; McIntosh, T.; Needham, D.; Evans, E. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys. J. 2000, 79, 328–339. [Google Scholar] [CrossRef]
- Lindahl, E.; Edholm, O. Mesoscopic undulations and thickness fluctuations in lipid bilayers from molecular dynamics simulations. Biophys. J. 2000, 79, 426–433. [Google Scholar] [CrossRef]
- Brandt, E.G.; Braun, A.R.; Sachs, J.N.; Nagle, J.F.; Edholm, O. Interpretation of fluctuation spectra in lipid bilayer simulations. Biophys. J. 2011, 100, 2104–2111. [Google Scholar] [CrossRef] [PubMed]
- Harmandaris, V.A.; Deserno, M. A novel method for measuring the bending rigidity of model lipid membranes by simulating tethers. J. Chem. Phys. 2006, 125, 204905:1–204905:6. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, R.F.; Fedorov, A.; Prieto, M. Sphingomyelin/phosphatidylcholine/cholesterol phase diagram: Boundaries and composition of lipid rafts. Biophys. J. 2003, 85, 2406–2416. [Google Scholar] [CrossRef]
- McMullen, T.P.; Lewis, R.N.; McElhaney, R.N. Cholesterol-phospholipid interactions, the liquid-ordered phase and lipid rafts in model and biological membranes. Curr. Opin. Colloid Interface Sci. 2004, 8, 459–468. [Google Scholar] [CrossRef]
- Arkhipov, A.; Yin, Y.; Schulten, K. Four-scale description of membrane sculpting by BAR domains. Biophys. J. 2008, 95, 2806–2821. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Arkhipov, A.; Schulten, K. Simulations of membrane tubulation by lattices of amphiphysin N-BAR domains. Structure 2009, 17, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Louhivuori, M.; Risselada, H.J.; van der Giessen, E.; Marrink, S.J. Release of content through mechano-sensitive gates in pressurized liposomes. Proc. Natl. Acad. Sci. USA 2010, 107, 19856–19860. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; Kuriyan, J. Molecular dynamics and protein function. Proc. Natl. Acad. Sci. USA 2005, 102, 6679–6685. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Van Gunsteren, W.F.; Dolenc, J.; Mark, A.E. Molecular simulation as an aid to experimentalists. Curr. Opin. Struct. Biol. 2008, 18, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Gipson, B.; Hsu, D.; Kavraki, L.E.; Latombe, J.C. Computational models of protein kinematics and dynamics: Beyond simulation. Annu. Rev. Anal. Chem. 2012, 5, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Lezon, T.R.; Yang, L.W.; Eyal, E. Global dynamics of proteins: Bridging between structure and function. Annu. Rev. Biophys. 2010, 39, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Adcock, S.; McCammon, J. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O.; MacKerell, Alexander D., Jr. Comparison of Protein Force Fields for Molecular Dynamics Simulations. In Methods Molecular Biology; Kukol, A., Ed.; Humana Press: New York, NY, USA, 2008; Volume 443, pp. 63–88. [Google Scholar]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D. How robust are protein folding simulations with respect to force field parameterization? Biophys. J. 2011, 100, L47–L49. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Systematic validation of protein force fields against experimental data. PLoS One 2012, 7, e32131. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, K.A.; Lin, Y.S.; Das, R.; Pande, V.S. Are protein force fields getting better? A systematic benchmark on 524 diverse NMR measurements. J. Chem. Theory Comput. 2012, 8, 1409–1414. [Google Scholar] [CrossRef] [PubMed]
- Snow, C.D.; Nguyen, H.; Pande, V.S.; Gruebele, M. Absolute comparison of simulated and experimental protein-folding dynamics. Nature 2002, 420, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; MacCallum, J.L. The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Cellmer, T.; Buscaglia, M.; Henry, E.R.; Hofrichter, J.; Eaton, W.A. Making connections between ultrafast protein folding kinetics and molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2011, 108, 6103–6108. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Gorfe, A.A.; McCammon, J.A. Large conformational changes in proteins: Signaling and other functions. Curr. Opin. Struct. Biol. 2010, 20, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Scheraga, H.A.; Khalili, M.; Liwo, A. Protein-folding dynamics: Overview of molecular simulation techniques. Annu. Rev. Phys. Chem. 2007, 58, 57–83. [Google Scholar] [CrossRef] [PubMed]
- Werner, T.; Morris, M.B.; Dastmalchi, S.; Church, W.B. Structural modelling and dynamics of proteins for insights into drug interactions. Adv. Drug Deliv. Rev. 2012, 64, 323–343. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking: Current status and future challenges. Proteins 2006, 65, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Lill, M.A. Efficient incorporation of protein flexibility and dynamics into molecular docking simulations. Biochemistry 2011, 50, 6157–6169. [Google Scholar] [CrossRef] [PubMed]
- Salsbury, F.R., Jr. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr. Opin. Pharmacol. 2010, 10, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Livesay, D.R. Protein dynamics: Dancing on an ever-changing free energy stage. Curr. Opin. Pharmacol. 2010, 10, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Singh Sandhu, K.; Hazai, E.; Hoksza, Z.; Kiss, H.J.M.; Miozzo, F.; Veres, D.V.; Piazza, F.; Nussinov, R. Disordered proteins and network disorder in network descriptions of protein structure, dynamics and function: Hypotheses and a comprehensive review. Curr. Protein Pept. Sci. 2011, 13, 19–33. [Google Scholar] [CrossRef]
- Chodera, J.D.; Mobley, D.L.; Shirts, M.R.; Dixon, R.W.; Branson, K.; Pande, V.S. Alchemical free energy methods for drug discovery: Progress and challenges. Curr. Opin. Struct. Biol. 2011, 21, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cao, Y. Protein mechanics: From single molecules to functional biomaterials. Acc. Chem. Res. 2010, 43, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Hsin, J.; Sotomayor, M.; Comellas, G.; Schulten, K. Discovery through the computational microscope. Structure 2009, 17, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Buch, I.; Giorgino, T.; de Fabritiis, G. Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2011, 108, 10184–10189. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.I.; Moss, D.J.; Knotts, T.A. Multiple molecule effects on the cooperativity of protein folding transitions in simulations. J. Chem. Phys. 2012, 136, 245101:1–245101:10. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Hsin, J.; Liu, Y.; Selvin, P.R.; Schulten, K. Formation of salt bridges mediates internal dimerization of myosin VI medial tail domain. Structure 2010, 18, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Lucent, D.; Snow, C.D.; Aitken, C.E.; Pande, V.S. Non-bulk-like solvent behavior in the ribosome exit tunnel. PLoS Comput. Biol. 2010, 6, e1000963. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.A.; Bradley, P. Atomistic modeling of protein-DNA interaction specificity: Progress and applications. Curr. Opin. Struct. Biol. 2012, 22, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Laughton, C.A.; Harris, S.A. The atomistic simulation of DNA. WIREs Comput. Mol. Sci. 2011, 1, 590–600. [Google Scholar] [CrossRef]
- Periole, X.; Cavalli, M.; Marrink, S.J.; Ceruso, M.A. Combining an elastic network with a coarse-grained molecular force field: Structure, dynamics, and intermolecular recognition. J. Chem. Theory Comput. 2009, 5, 2531–2543. [Google Scholar] [CrossRef]
- Zhang, Z.; Pfaendtner, J.; Grafmüller, A.; Voth, G.A. Defining coarse-grained representations of large biomolecules and biomolecular complexes from elastic network models. Biophys. J. 2009, 97, 2327–2337. [Google Scholar] [CrossRef] [PubMed]
- Lyman, E.; Pfaendtner, J.; Voth, G. Systematic multiscale parameterization of heterogeneous elastic network models of proteins. Biophys. J. 2008, 95, 4183–4192. [Google Scholar] [CrossRef] [PubMed]
- Zwier, M.C.; Chong, L.T. Reaching biological timescales with all-atom molecular dynamics simulations. Curr. Opin. Pharmacol. 2010, 10, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Genchev, G.Z.; Källberg, M.; Gürsoy, G.; Mittal, A.; Dubey, L.; Perisic, O.; Feng, G.; Langlois, R.; Lu, H. Mechanical signaling on the single protein level studied using steered molecular dynamics. Cell Biochem. Biophys. 2009, 55, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71. [Google Scholar] [CrossRef]
- Maragliano, L.; Vanden-Eijnden, E. A temperature accelerated method for sampling free energy and determining reaction pathways in rare events simulations. Chem. Phys. Lett. 2006, 426, 168–175. [Google Scholar] [CrossRef]
- De Ruiter, A.; Oostenbrink, C. Free energy calculations of protein-ligand interactions. Curr. Opin. Chem. Biol. 2011, 15, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, S.; Staneva, I.; Mohanty, S.; Irbäck, A. Monte carlo studies of protein aggregation. Phys. Procedia 2012, 34, 49–54. [Google Scholar] [CrossRef]
- Ribeiro, A.A.; de Alencastro, R.B. Mixed monte carlo-molecular dynamics simulations of the prion protein. J. Mol. Graphics Modell. 2013, 42, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sun, Y. Molecular simulation of adsorption and its implications to protein chromatography: A review. Biochem. Eng. J. 2010, 48, 408–415. [Google Scholar] [CrossRef]
- Sapienza, P.J.; Lee, A.L. Using NMR to study fast dynamics in proteins: Methods and applications. Curr. Opin. Pharmacol. 2010, 10, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bouguet-Bonnet, S.; Buck, M. Combining NMR and Molecular Dynamics Studies for Insights into the Allostery of Small GTPase-Protein Interactions. In Methods in Molecular Biology; Fenton, A.W., Ed.; Springer: New York, NY, USA, 2012; Volume 796, pp. 235–259. [Google Scholar]
- Shell, M.S. The relative entropy is fundamental to multiscale and inverse thermodynamic problems. J. Chem. Phys. 2008, 129, 144108:1–144108:7. [Google Scholar] [CrossRef] [PubMed]
- Ash, W.L.; Zlomislic, M.R.; Oloo, E.O.; Tieleman, D.P. Computer simulations of membrane proteins. Biochim. Biophys. Acta Biomembr. 2004, 1666, 158–189. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, E.; Sansom, M.S. Membrane proteins: Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2008, 18, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Sadiq, S.; Guixa-Gonzalez, R.; Dainese, E.; Pastor, M.; de Fabritiis, G.; Selent, J. Molecular modeling and simulation of membrane lipid-mediated effects on GPCRs. Curr. Med. Chem. 2013, 20, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Kindt, J.T. Atomistic simulation of hydrophobic matching effects on lipid composition near a helical peptide embedded in mixed-lipid bilayers. J. Phys. Chem. B 2010, 114, 8076–8080. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, J.D.; Drasler, W.J.; Xie, W.; Gao, J.; Popot, J.L.; Sachs, J.N. All-atom and coarse-grained molecular dynamics simulations of a membrane protein stabilizing polymer. Langmuir 2011, 27, 10523–10537. [Google Scholar] [CrossRef] [PubMed]
- Wee, C.L.; Balali-Mood, K.; Gavaghan, D.; Sansom, M.S. The interaction of phospholipase A2 with a phospholipid bilayer: Coarse-grained molecular dynamics simulations. Biophys. J. 2008, 95, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Thøgersen, L.; Schiøtt, B.; Vosegaard, T.; Nielsen, N.C.; Tajkhorshid, E. Peptide aggregation and pore formation in a lipid bilayer: A combined coarse-grained and all atom molecular dynamics study. Biophys. J. 2008, 95, 4337–4347. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Peter, C.; van der Vegt, N.F.A. Self-assembling dipeptides: Conformational sampling in solvent-free coarse-grained simulation. Phys. Chem. Chem. Phys. 2009, 11, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Rzepiela, A.J.; Schafer, L.V.; Goga, N.; Risselada, H.J.; de Vries, A.H.; Marrink, S.J. Reconstruction of atomistic details from coarse-grained structures. J. Comput. Chem. 2010, 31, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Bezkorovaynaya, O.; Lukyanov, A.; Kremer, K.; Peter, C. Multiscale simulation of small peptides: Consistent conformational sampling in atomistic and coarse-grained models. J. Comput. Chem. 2012, 33, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y.; Wolynes, P.G.; Onuchic, J.N. Protein topology determines binding mechanism. Proc. Natl. Acad. Sci. USA 2004, 101, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Hall, C.K. Molecular dynamics simulations of spontaneous fibril formation by random-coil peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 16180–16185. [Google Scholar] [CrossRef] [PubMed]
- Emperador, A.; Carrillo, O.; Rueda, M.; Orozco, M. Exploring the suitability of coarse-grained techniques for the representation of protein dynamics. Biophys. J. 2008, 95, 2127–2138. [Google Scholar] [CrossRef] [PubMed]
- Elcock, A.H. Atomic-level observation of macromolecular crowding effects: Escape of a protein from the GroEL cage. Proc. Natl. Acad. Sci. USA 2003, 100, 2340–2344. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Rader, A. Coarse-grained normal mode analysis in structural biology. Curr. Opin. Struct. Biol. 2005, 15, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Head-Gordon, T.; Brown, S. Minimalist models for protein folding and design. Curr. Opin. Struct. Biol. 2003, 13, 160–167. [Google Scholar] [CrossRef]
- Coluzza, I. A coarse-grained approach to protein design: Learning from design to understand folding. PLoS One 2011, 6, e20853. [Google Scholar] [CrossRef] [PubMed]
- DeVane, R.; Shinoda, W.; Moore, P.B.; Klein, M.L. Transferable coarse grain nonbonded interaction model for amino acids. J. Chem. Theory Comput. 2009, 5, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Wan, C.K.; Wu, Y.D. Toward a coarse-grained protein model coupled with a coarse-grained solvent model: Solvation free energies of amino acid side chains. J. Chem. Theory Comput. 2008, 4, 1891–1901. [Google Scholar] [CrossRef]
- Basdevant, N.; Borgis, D.; Ha-Duong, T. A coarse-grained protein-protein potential derived from an all-atom force field. J. Phys. Chem. B 2007, 111, 9390–9399. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Arkhipov, A.; Freddolino, P.L.; Schulten, K. Coarse grained protein-lipid model with application to lipoprotein particles. J. Phys. Chem. B 2006, 110, 3674–3684. [Google Scholar] [CrossRef] [PubMed]
- Freddolino, P.L.; Shih, A.Y.; Arkhipov, A.; Ying, Y.; Chen, Z.; Schulten, K. Application of Residue-Based and Shape-Based Coarse-Graining to Biomolecular Simulations. In Coarse-Graining of Condensed Phase and Biomolecular Systems; CRC Press: Boca Raton, FL, USA, 2009; p. 299. [Google Scholar]
- Mim, C.; Cui, H.; Gawronski-Salerno, J.; Frost, A.; Lyman, E.; Voth, G.; Unger, V. Structural basis of membrane bending by the N-BAR protein endophilin. Cell 2012, 149, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Voth, G.A. Hybrid approach for highly coarse-grained lipid bilayer models. J. Chem. Theory Comput. 2012, 9, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.M.; Mukherjee, S.; Cheng, Y.M.; Feig, M. PRIMO/PRIMONA: A coarse-grained model for proteins and nucleic acids that preserves near-atomistic accuracy. Proteins 2010, 78, 1266–1281. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.J. The MARTINI coarse-grained force field: Extension to proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef]
- Radzicka, A.; Wolfenden, R. Comparing the polarities of the amino acids: Side-chain distribution coefficients between the vapor phase, cyclohexane, 1-octanol, and neutral aqueous solution. Biochemistry 1988, 27, 1664–1670. [Google Scholar] [CrossRef]
- Wolfenden, R.; Andersson, L.; Cullis, P.M.; Southgate, C.C.B. Affinities of amino acid side chains for solvent water. Biochemistry 1981, 20, 849–855. [Google Scholar] [CrossRef] [PubMed]
- De Jong, D.H.; Singh, G.; Bennett, W.F.D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved parameters for the martini coarse-grained protein force field. J. Chem. Theory Comput. 2012, 9, 687–697. [Google Scholar] [CrossRef]
- Shirts, M.; Chodera, J. Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys. 2008, 129, 124105:1–124105:10. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- MacCallum, J.L.; Bennett, W.F.D.; Tieleman, D.P. Distribution of amino acids in a lipid bilayer from computer simulations. Biophys. J. 2008, 94, 3393–3404. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Tieleman, D.P. Using the wimley-white hydrophobicity scale as a direct quantitative test of force fields: The MARTINI coarse-grained model. J. Chem. Theory Comput. 2011, 7, 2316–2324. [Google Scholar] [CrossRef]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Mol. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef]
- De Jong, D.H.; Periole, X.; Marrink, S.J. Dimerization of amino acid side chains: Lessons from the comparison of different force fields. J. Chem. Theory Comput. 2012, 8, 1003–1014. [Google Scholar] [CrossRef]
- Miyazawa, S.; Jernigan, R.L. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J. Mol. Biol. 1996, 256, 623–644. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.; Rauscher, S.; Pomès, R.; Tieleman, D.P. Improving internal peptide dynamics in the coarse-grained MARTINI model: Toward large-scale simulations of amyloid- and elastin-like peptides. J. Chem. Theory Comput. 2012, 8, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Faller, R.; Marrink, S.J. Simulation of domain formation in mixed DLPC-DSPC bilayers. Langmuir 2004, 20, 7686–7693. [Google Scholar] [CrossRef] [PubMed]
- Rand, R.; Fuller, N. Structural dimensions and their changes in a reentrant hexagonal-lamellar transition of phospholipids. Biophys. J. 1994, 66, 2127–2138. [Google Scholar] [CrossRef]
- Yang, L.; Ding, L.; Huang, H.W. New phases of phospholipids and implications to the membrane fusion problem. Biochemistry 2003, 42, 6631–6635. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Mark, A.E. Molecular view of hexagonal phase formation in phospholipid membranes. Biophys. J. 2004, 87, 3894–3900. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Risselada, J.; Mark, A.E. Simulation of gel phase formation and melting in lipid bilayers using a coarse grained model. Chem. Phys. Lipids 2005, 135, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Squires, A.M.; Conn, C.E.; Ces, O.; Heron, A.J.; Mulet, X.; Shearman, G.C.; Templer, R.H. Pressure-jump X-ray studies of liquid crystal transitions in lipids. Philos. Trans. R. Soc. Lond. A 2006, 364, 2635–2655. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, L.V.; Marrink, S.J. Partitioning of lipids at domain boundaries in model membranes. Biophys. J. 2010, 99, L91–L93. [Google Scholar] [CrossRef] [PubMed]
- Khalfa, A.; Tarek, M. On the antibacterial action of cyclic peptides: Insights from coarse-grained MD simulations. J. Phys. Chem. B 2010, 114, 2676–2684. [Google Scholar] [CrossRef] [PubMed]
- Polyansky, A.A.; Ramaswamy, R.; Volynsky, P.E.; Sbalzarini, I.F.; Marrink, S.J.; Efremov, R.G. Antimicrobial peptides induce growth of phosphatidylglycerol domains in a model bacterial membrane. J. Phys. Chem. Lett. 2010, 1, 3108–3111. [Google Scholar] [CrossRef]
- Kasson, P.M.; Kelley, N.W.; Singhal, N.; Vrljic, M.; Brunger, A.T.; Pande, V.S. Ensemble molecular dynamics yields submillisecond kinetics and intermediates of membrane fusion. Proc. Natl. Acad. Sci. USA 2006, 103, 11916–11921. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Mark, A.E. The mechanism of vesicle fusion as revealed by molecular dynamics simulations. J. Am. Chem. Soc. 2003, 125, 11144–11145. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, Y.G.; Marrink, S.J.; Lipowsky, R.; Knecht, V. Solvent-exposed tails as prestalk transition states for membrane fusion at low hydration. J. Am. Chem. Soc. 2010, 132, 6710–6718. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.L.; Larson, R.G. Folding of lipid monolayers containing lung surfactant proteins SP-B1-25 and SP-C studied via coarse-grained molecular dynamics simulations. Biochim. Biophys. Acta Biomembr. 2010, 1798, 1632–1650. [Google Scholar] [CrossRef] [PubMed]
- Corsi, J.; Hawtin, R.W.; Ces, O.; Attard, G.S.; Khalid, S. DNA lipoplexes: Formation of the inverse hexagonal phase observed by coarse-grained molecular dynamics simulation. Langmuir 2010, 26, 12119–12125. [Google Scholar] [CrossRef] [PubMed]
- Pantano, D.A.; Moore, P.B.; Klein, M.L.; Discher, D.E. Raft registration across bilayers in a molecularly detailed model. Soft Matter 2011, 7, 8182–8191. [Google Scholar] [CrossRef]
- Risselada, H.J.; Marrink, S.J. The molecular face of lipid rafts in model membranes. Proc. Natl. Acad. Sci. USA 2008, 105, 17367–17372. [Google Scholar] [CrossRef] [PubMed]
- Cooke, I.R.; Kremer, K.; Deserno, M. Tunable generic model for fluid bilayer membranes. Phys. Rev. E 2005, 72, 011506:1–011506:4. [Google Scholar] [CrossRef]
- Reynwar, B.J.; Illya, G.; Harmandaris, V.A.; Muller, M.M.; Kremer, K.; Deserno, M. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature 2007, 447, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Brannigan, G.; Lin, L.L.; Brown, F. Implicit solvent simulation models for biomembranes. Eur. Biophys. J. 2006, 35, 104–124. [Google Scholar] [CrossRef] [PubMed]
- West, B.; Brown, F.L.; Schmid, F. Membrane-protein interactions in a generic coarse-grained model for lipid bilayers. Biophys. J. 2009, 96, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Sodt, A.J.; Head-Gordon, T. An implicit solvent coarse-grained lipid model with correct stress profile. J. Chem. Phys. 2010, 132, 205103:1–205103:8. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Mim, C.; Vázquez, F.; Lyman, E.; Unger, V.; Voth, G. Understanding the role of amphipathic helices in N-BAR domain driven membrane remodeling. Biophys. J. 2013, 104, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Ford, M.G.J.; Mills, I.G.; Peter, B.J.; Vallis, Y.; Praefcke, G.J.K.; Evans, P.R.; McMahon, H.T. Curvature of clathrin-coated pits driven by epsin. Nature 2002, 419, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Kweon, D.; Shin, Y.; Shin, J.; Lee, J.; Lee, J.; Seo, J.; Kim, Y. Membrane topology of helix 0 of the Epsin N-terminal homology domain. Mol. Cells 2006, 21, 428–435. [Google Scholar] [PubMed]
- Lai, C.L.; Jao, C.C.; Lyman, E.; Gallop, J.L.; Peter, B.J.; McMahon, H.T.; Langen, R.; Voth, G.A. Membrane binding and self-association of the epsin n-terminal homology domain. J. Mol. Biol. 2012, 423, 800–817. [Google Scholar] [CrossRef] [PubMed]
- Braun, A.R.; Sevcsik, E.; Chin, P.; Rhoades, E.; Tristram-Nagle, S.; Sachs, J.N. α-synuclein induces both positive mean curvature and negative gaussian curvature in membranes. J. Am. Chem. Soc. 2012, 134, 2613–2620. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, T.; Capraro, B.R.; Zhu, C.; Das, S.L. Thermodynamics and mechanics of membrane curvature generation and sensing by proteins and lipids. Annu. Rev. Phys. Chem. 2011, 62, 483–506. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, G.; Klein, M.L. Computational approaches to nanobiotechnology: Probing the interaction of synthetic molecules with phospholipid bilayers via a coarse grain model. Nanotechnology 2004, 15. [Google Scholar] [CrossRef]
- Nielsen, S.O.; Lopez, C.F.; Ivanov, I.; Moore, P.B.; Shelley, J.C.; Klein, M.L. Transmembrane peptide-induced lipid sorting and mechanism of Lα-to-inverted phase transition using coarse-grain molecular dynamics. Biophys. J. 2004, 87, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.O.; Ensing, B.; Ortiz, V.; Moore, P.B.; Klein, M.L. Lipid bilayer perturbations around a transmembrane nanotube: A coarse grain molecular dynamics study. Biophys. J. 2005, 88, 3822–3828. [Google Scholar] [CrossRef] [PubMed]
- Cheung, D.L. Molecular simulation of hydrophobin adsorption at an oil-water interface. Langmuir 2012, 28, 8730–8736. [Google Scholar] [CrossRef] [PubMed]
- Ramadurai, S.; Holt, A.; Schäfer, L.V.; Krasnikov, V.V.; Rijkers, D.T.; Marrink, S.J.; Killian, J.A.; Poolman, B. Influence of hydrophobic mismatch and amino acid composition on the lateral diffusion of transmembrane peptides. Biophys. J. 2010, 99, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Ayton, G.S.; Voth, G.A. Multiscale computer simulation of the immature HIV-1 virion. Biophys. J. 2010, 99, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, F.J.M.; Venturoli, M.; Smit, B. Molecular simulations of lipid-mediated protein-protein interactions. Biophys. J. 2008, 95, 1851–1865. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, F.J.M.; Rodgers, J.M.; Willems, T.F.; Smit, B. Molecular simulation of the effect of cholesterol on lipid-mediated protein-protein interactions. Biophys. J. 2010, 99, 3629–3638. [Google Scholar] [CrossRef] [PubMed]
- Treptow, W.; Marrink, S.J.; Tarek, M. Gating motions in voltage-gated potassium channels revealed by coarse-grained molecular dynamics simulations. J. Phys. Chem. B 2008, 112, 3277–3282. [Google Scholar] [CrossRef] [PubMed]
- Wallace, E.J.; Sansom, M.S.P. Blocking of carbon nanotube based nanoinjectors by lipids: A simulation study. Nano Lett. 2008, 8, 2751–2756. [Google Scholar] [CrossRef] [PubMed]
- Wong-Ekkabut, J.; Baoukina, S.; Triampo, W.; Tang, I.M.; Tieleman, D.P.; Monticelli, L. Computer simulation study of fullerene translocation through lipid membranes. Nat. Nano 2008, 3, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, M.; Faller, R. Coarse-grained simulations of ABA amphiphilic triblock copolymer solutions in thin films. Phys. Chem. Chem. Phys. 2007, 9, 4662–4672. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Larson, R.G. Molecular dynamics simulations of PAMAM dendrimer-induced pore formation in DPPC bilayers with a coarse-grained model. J. Phys. Chem. B 2006, 110, 18204–18211. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Larson, R.G. Coarse-grained molecular dynamics studies of the concentration and size dependence of fifth- and seventh-generation PAMAM dendrimers on pore formation in DMPC bilayer. J. Phys. Chem. B 2008, 112, 7778–7784. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Larson, R.G. Lipid bilayer curvature and pore formation induced by charged linear polymers and dendrimers: The effect of molecular shape. J. Phys. Chem. B 2008, 112, 12279–12285. [Google Scholar] [CrossRef] [PubMed]
- Periole, X.; Huber, T.; Marrink, S.J.; Sakmar, T.P. G protein-coupled receptors self-assemble in dynamics simulations of model bilayers. J. Am. Chem. Soc. 2007, 129, 10126–10132. [Google Scholar] [CrossRef] [PubMed]
- Bond, P.J.; Sansom, M.S.P. Insertion and assembly of membrane proteins via simulation. J. Am. Chem. Soc. 2006, 128, 2697–2704. [Google Scholar] [CrossRef] [PubMed]
- Bond, P.J.; Wee, C.L.; Sansom, M.S.P. Coarse-grained molecular dynamics simulations of the energetics of helix insertion into a lipid bilayer. Biochemistry 2008, 47, 11321–11331. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, K.R.; Fleming, K.G. Association energetics of membrane spanning α-helices. Curr. Opin. Struct. Biol. 2008, 18, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Vuorela, T.; Catte, A.; Niemelä, P.S.; Hall, A.; Hyvönen, M.T.; Marrink, S.J.; Karttunen, M.; Vattulainen, I. Role of lipids in spheroidal high density lipoproteins. PLoS Comput. Biol. 2010, 6, e1000964. [Google Scholar] [CrossRef] [PubMed]
- Neri, M.; Anselmi, C.; Cascella, M.; Maritan, A.; Carloni, P. Coarse-grained model of proteins incorporating atomistic detail of the active site. Phys. Rev. Lett. 2005, 95, 218102:1–218102:4. [Google Scholar] [CrossRef]
- Neri, M.; Anselmi, C.; Carnevale, V.; Vargiu, A.V.; Carloni, P. Molecular dynamics simulations of outer-membrane protease T from E. coli based on a hybrid coarse-grained/atomistic potential. J. Phys. Condens. Matter 2006, 18. [Google Scholar] [CrossRef]
- Leguèbe, M.; Nguyen, C.; Capece, L.; Hoang, Z.; Giorgetti, A.; Carloni, P. Hybrid molecular mechanics/coarse-grained simulations for structural prediction of G-protein coupled receptor/ligand complexes. PLoS One 2012, 7, e47332. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Im, W.; Brooks, C.L. Interfacial folding and membrane insertion of designed peptides studied by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2005, 102, 6771–6776. [Google Scholar] [CrossRef] [PubMed]
- Rzepiela, A.J.; Louhivuori, M.; Peter, C.; Marrink, S.J. Hybrid simulations: Combining atomistic and coarse-grained force fields using virtual sites. Phys. Chem. Chem. Phys. 2011, 13, 10437–10448. [Google Scholar] [CrossRef] [PubMed]
- Seifert, U. Configurations of fluid membranes and vesicles. Adv. Phys. 1997, 46, 13–137. [Google Scholar] [CrossRef]
- Helfrich, W. Effect of thermal undulations on the rigidity of fluid membranes and interfaces. J. Phys. Fr. 1985, 46, 1263–1268. [Google Scholar] [CrossRef]
- Föster, D. On the scale dependence, due to thermal fluctuations, of the elastic properties of membranes. Phys. Lett. A 1986, 114, 115–120. [Google Scholar] [CrossRef]
- Brannigan, G.; Brown, F.L. A consistent model for thermal fluctuations and protein-induced deformations in lipid bilayers. Biophys. J. 2006, 90, 1501–1520. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Yoo, J.; Yethiraj, A.; Cui, Q.; Chen, X. Mechanosensitive channels: Insights from continuum-based simulations. Cell Biochem. Biophys. 2008, 52, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ayton, G.S.; McWhirter, J.L.; Voth, G.A. A second generation mesoscopic lipid bilayer model: Connections to field-theory descriptions of membranes and nonlocal hydrodynamics. J. Chem. Phys. 2006, 124, 064906:1–064906:12. [Google Scholar] [CrossRef] [PubMed]
- Ayton, G.S.; McWhirter, J.L.; McMurtry, P.; Voth, G.A. Coupling field theory with continuum mechanics: A simulation of domain formation in giant unilamellar vesicles. Biophys. J. 2005, 88, 3855–3869. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Ayton, G.S.; Voth, G.A. Multiscale coupling of mesoscopic-and atomistic-level lipid bilayer simulations. J. Chem. Phys. 2005, 122, 244716:1–244716:12. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.J.; Radhakrishnan, R. Calculation of free energies in fluid membranes subject to heterogeneous curvature fields. Phys. Rev. E 2009, 80, 011925:1–011925:8. [Google Scholar] [CrossRef]
- Agrawal, N.J.; Nukpezah, J.; Radhakrishnan, R. Minimal mesoscale model for protein-mediated vesiculation in clathrin-dependent endocytosis. PLoS Comput. Biol. 2010, 6, e1000926. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tourdot, R.; Ramanan, V.; Agrawal, N.J.; Radhakrishanan, R. Mesoscale simulations of curvature-inducing protein partitioning on lipid bilayer membranes in the presence of mean curvature fields. Mol. Phys. 2012, 110, 1127–1137. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bradley, R.; Radhakrishnan, R. Coarse-Grained Models for Protein-Cell Membrane Interactions. Polymers 2013, 5, 890-936. https://doi.org/10.3390/polym5030890
Bradley R, Radhakrishnan R. Coarse-Grained Models for Protein-Cell Membrane Interactions. Polymers. 2013; 5(3):890-936. https://doi.org/10.3390/polym5030890
Chicago/Turabian StyleBradley, Ryan, and Ravi Radhakrishnan. 2013. "Coarse-Grained Models for Protein-Cell Membrane Interactions" Polymers 5, no. 3: 890-936. https://doi.org/10.3390/polym5030890
APA StyleBradley, R., & Radhakrishnan, R. (2013). Coarse-Grained Models for Protein-Cell Membrane Interactions. Polymers, 5(3), 890-936. https://doi.org/10.3390/polym5030890