Enzymatic Synthesis and Crosslinking of Novel High Molecular Weight Polyepoxyricinoleate

Abstract
:1. Introduction
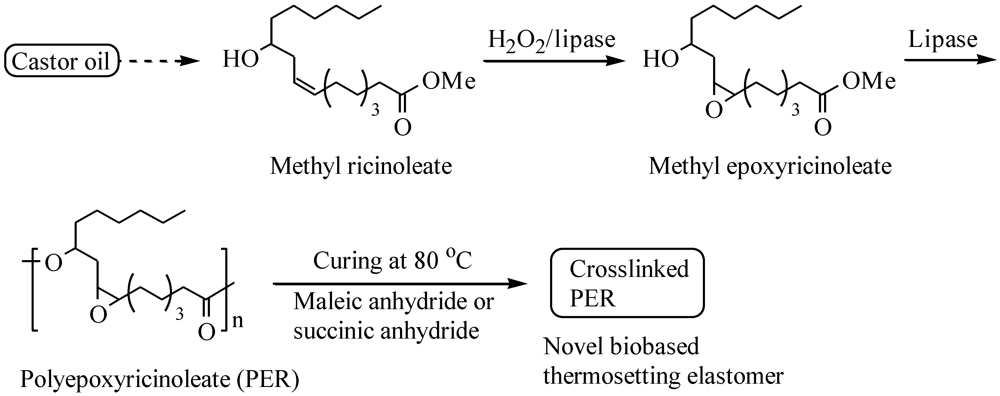
2. Experimental Section
2.1. Materials
2.2. Measurements
2.3. General Enzymatic Epoxidation Procedure
2.4. General Enzymatic Polymerization Procedure
2.5. General Crosslinking Procedure for PER
3. Results and Discussion
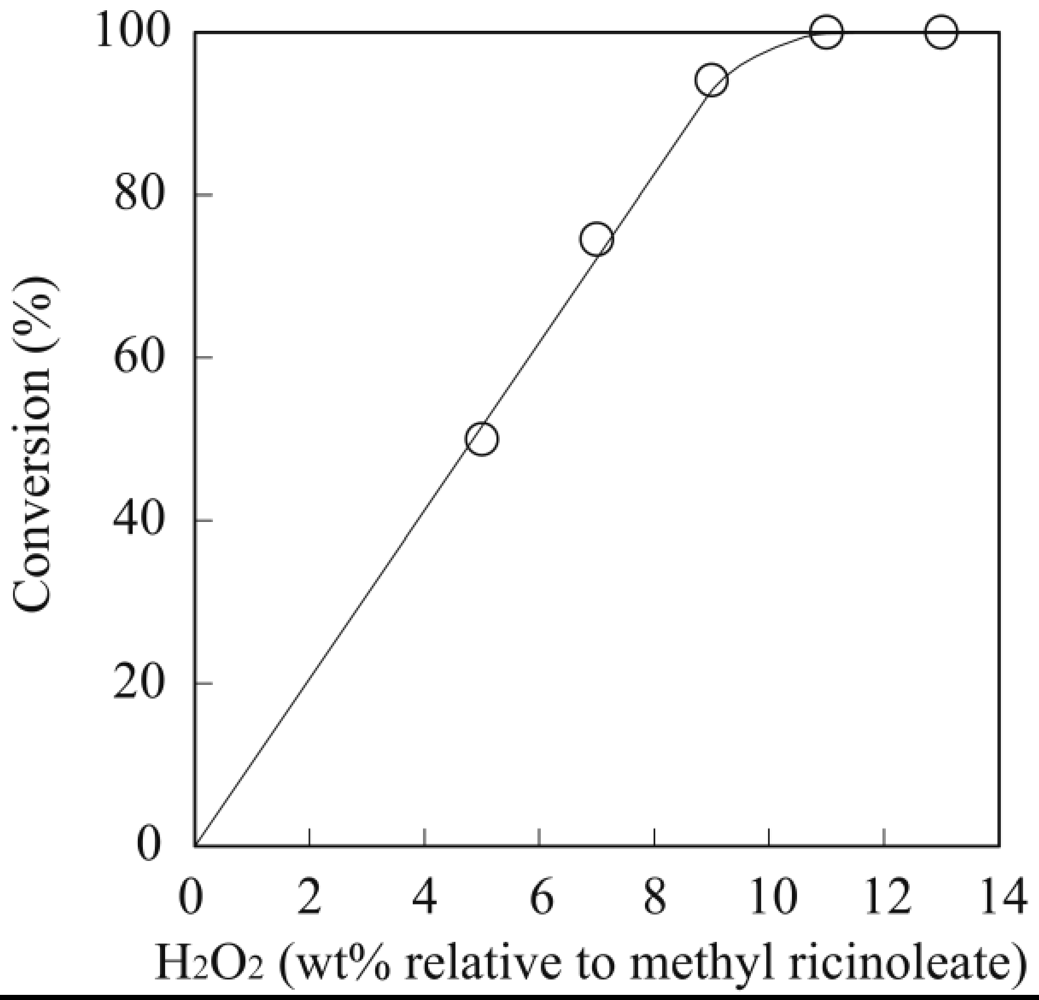
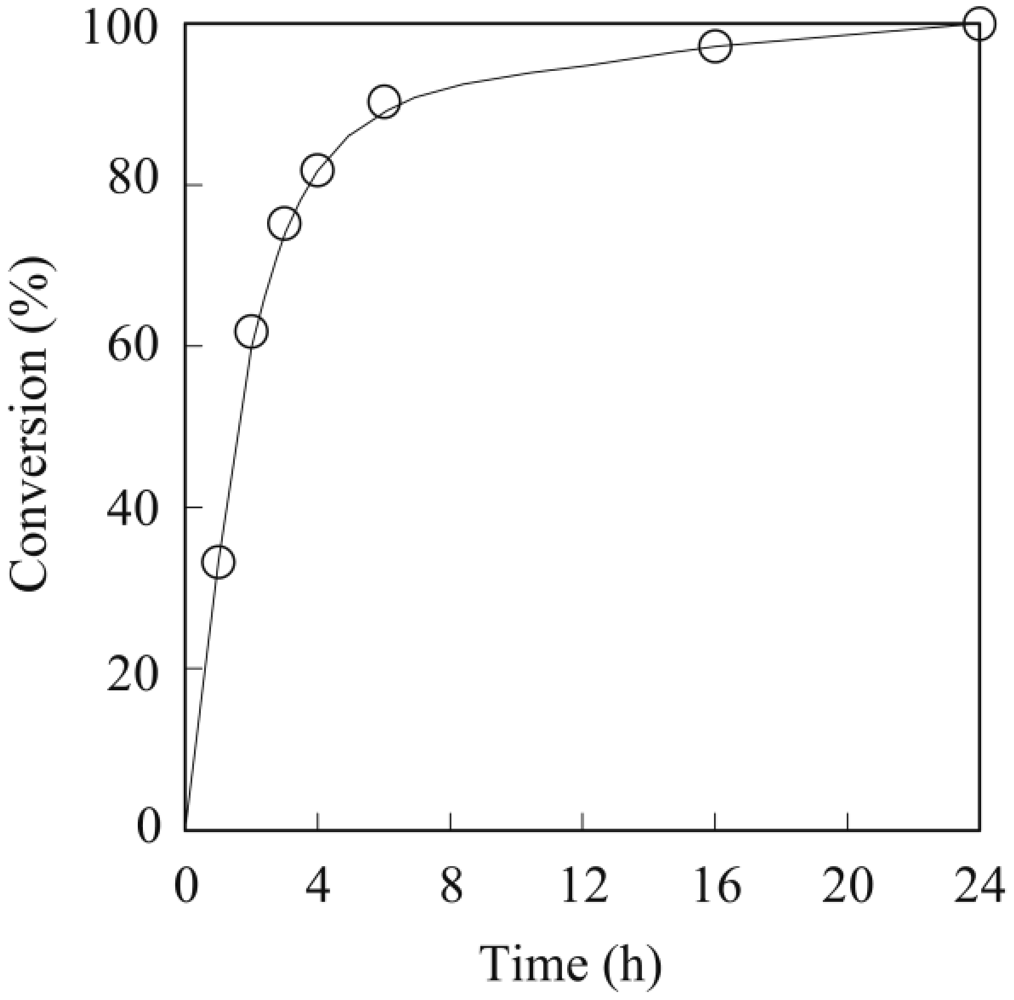
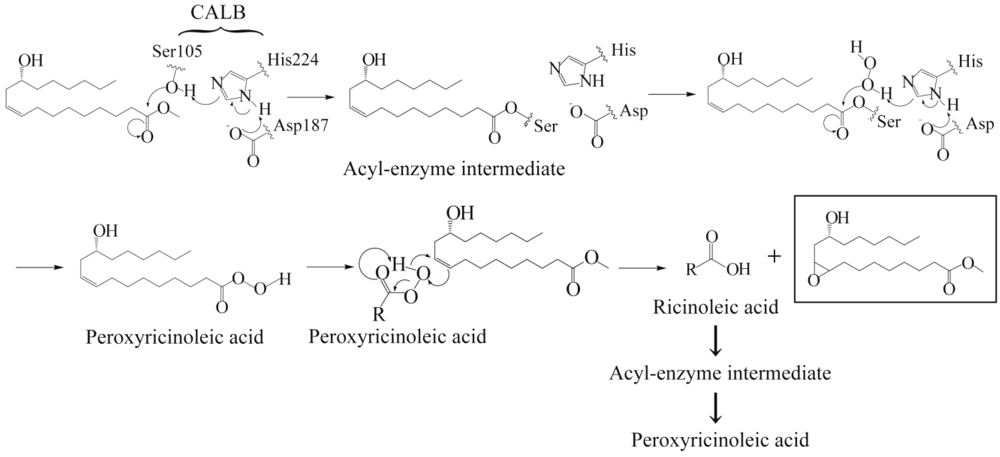
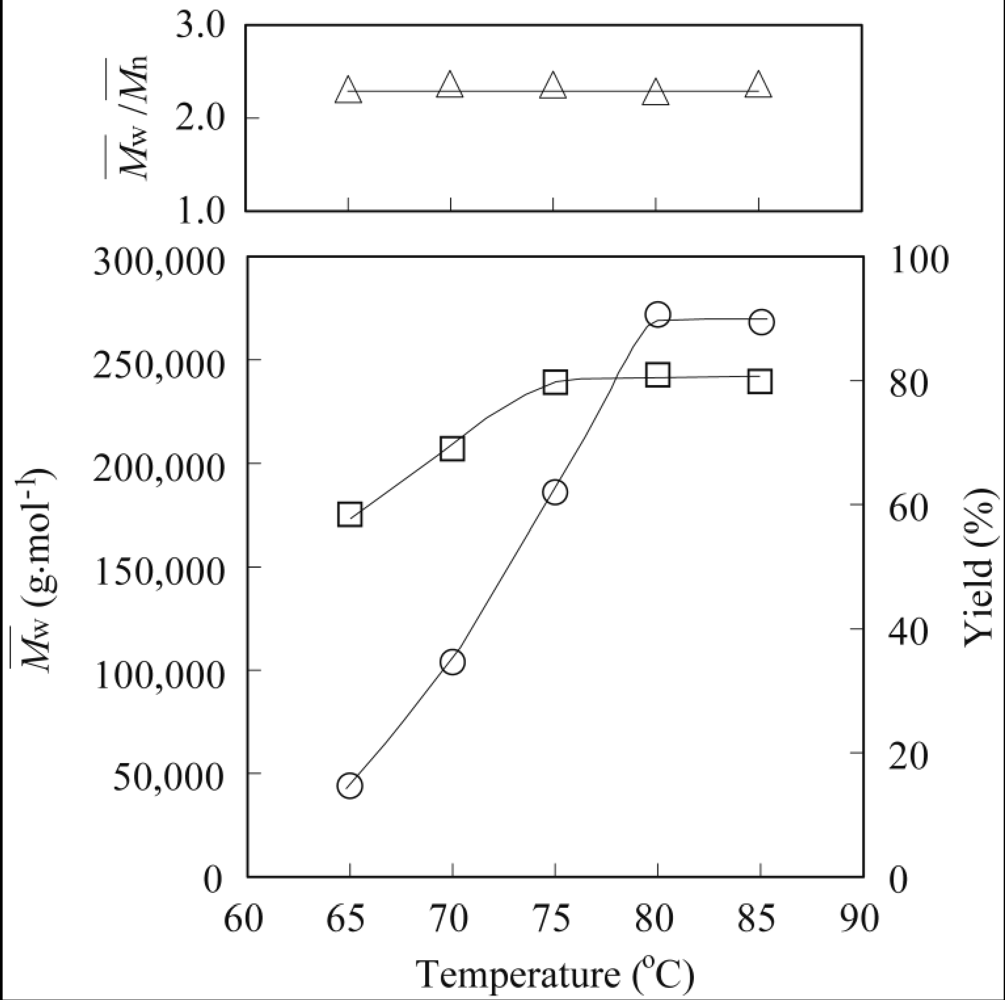
3.1. Lipase-Catalyzed Epoxidation of Methyl Ricinoleate
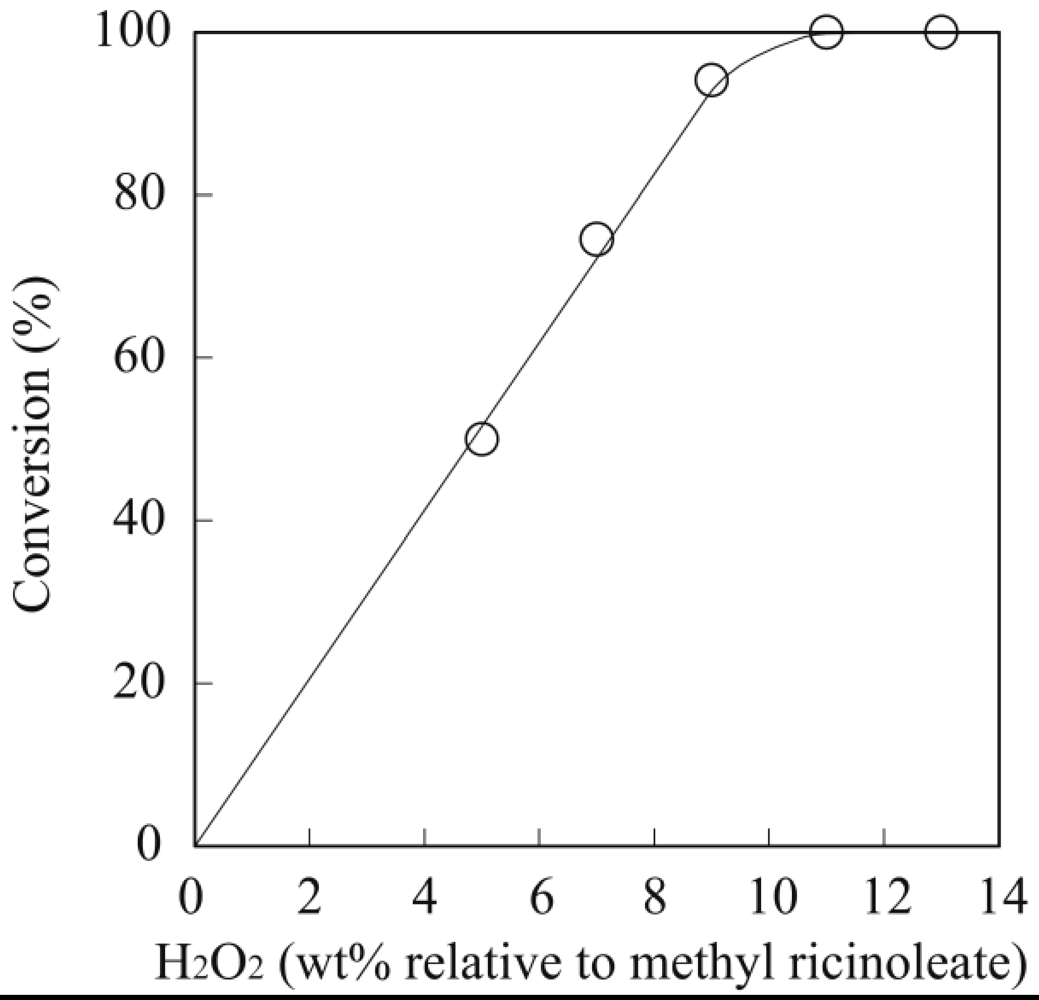
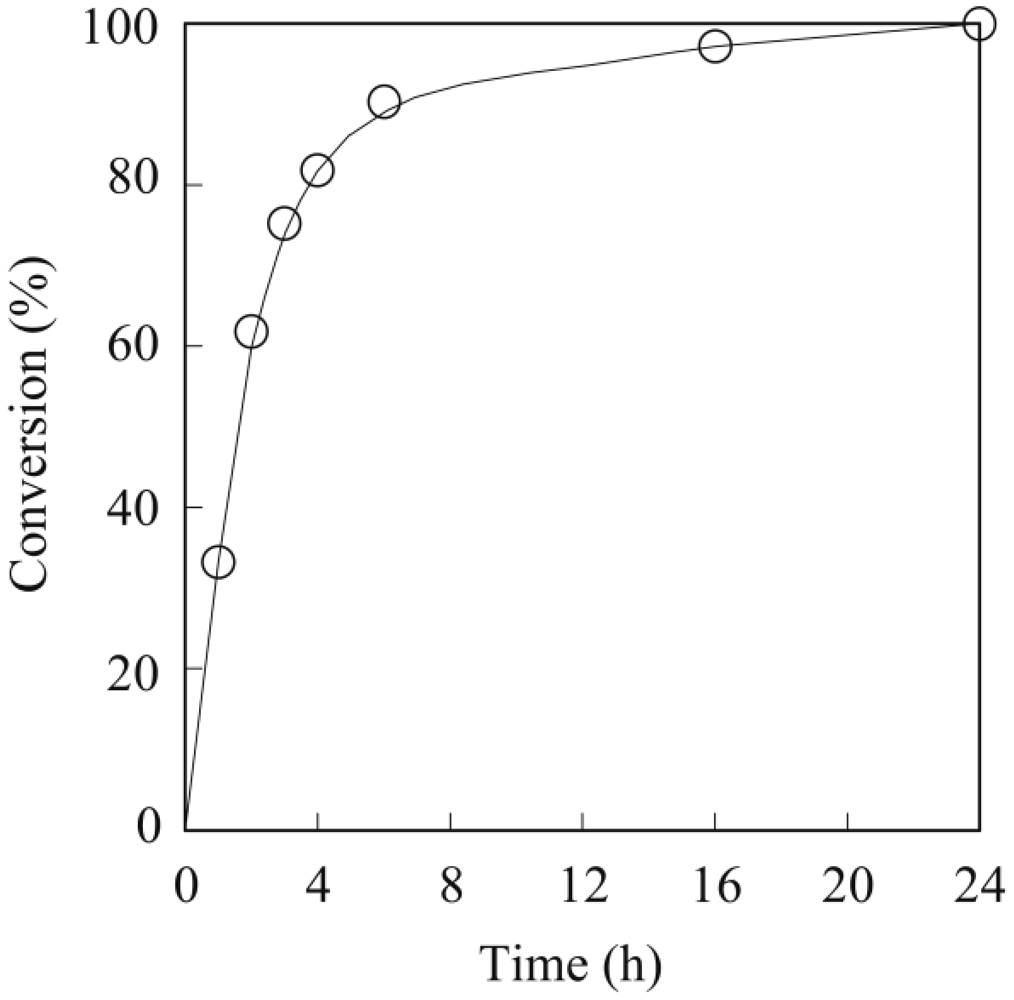
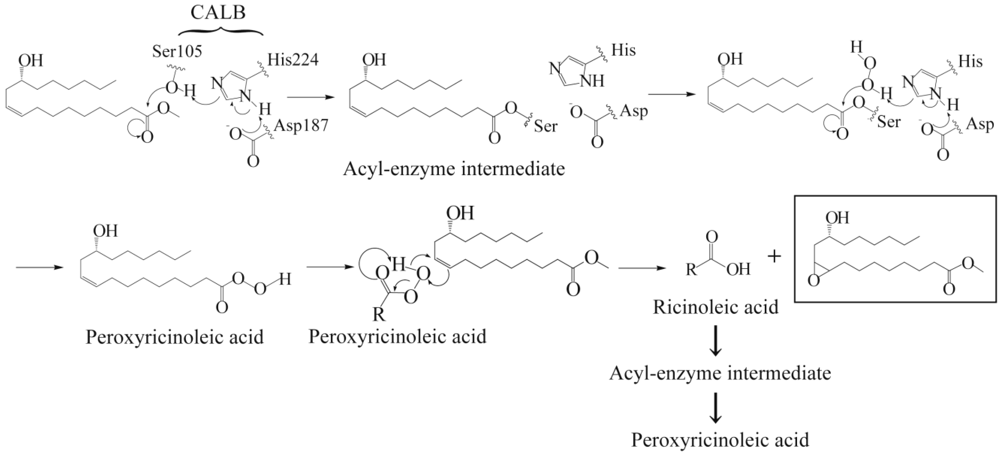
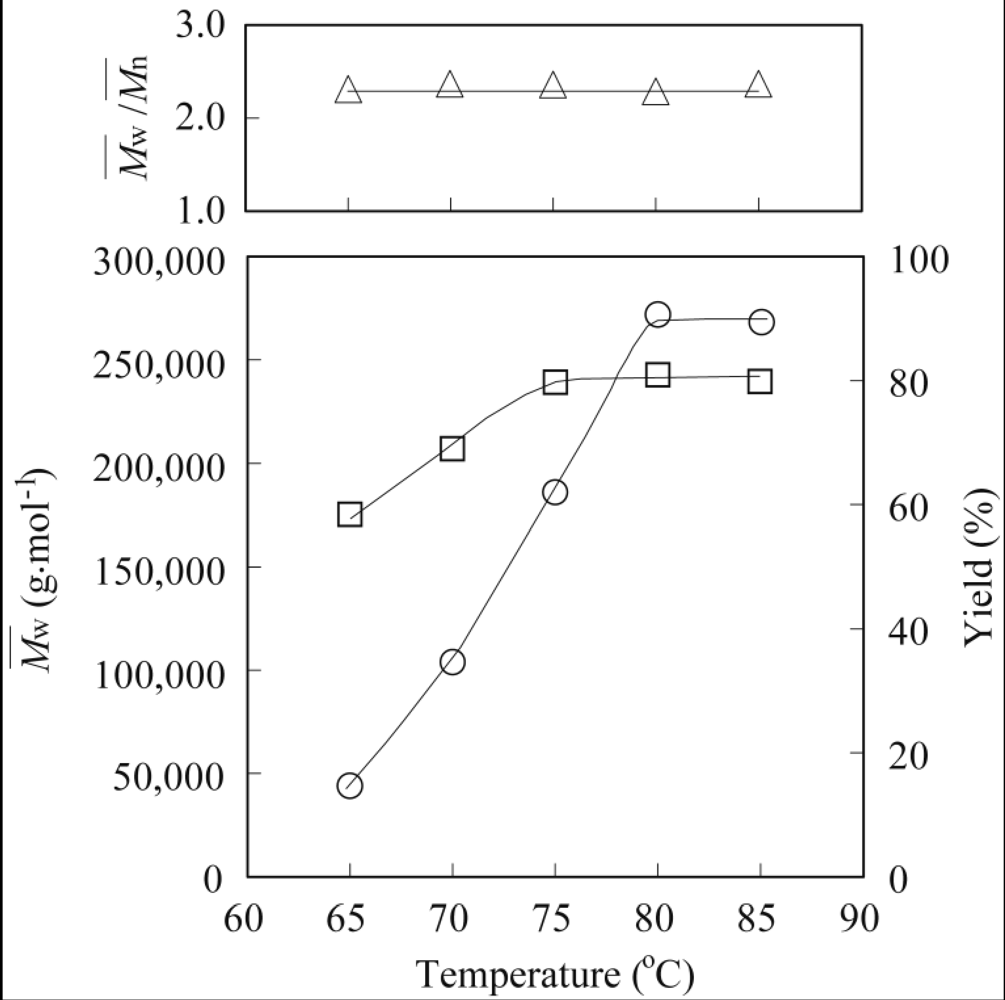
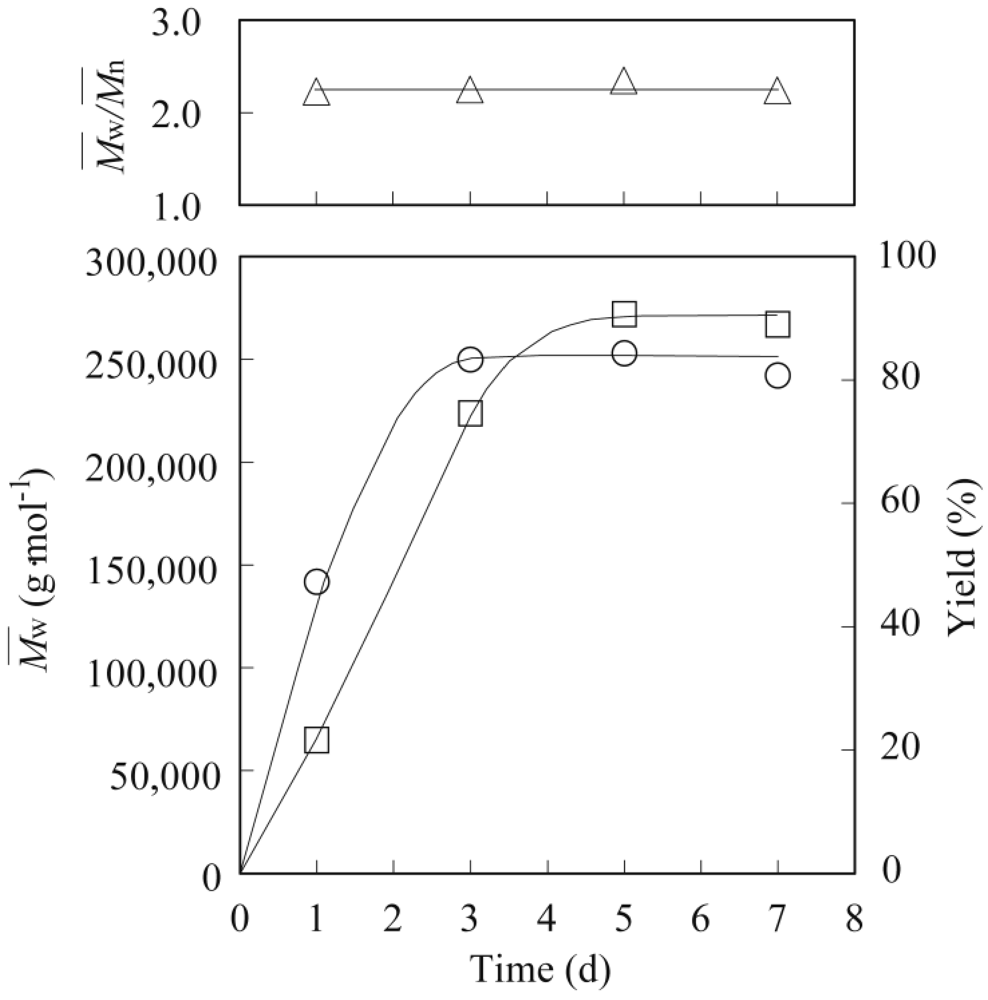
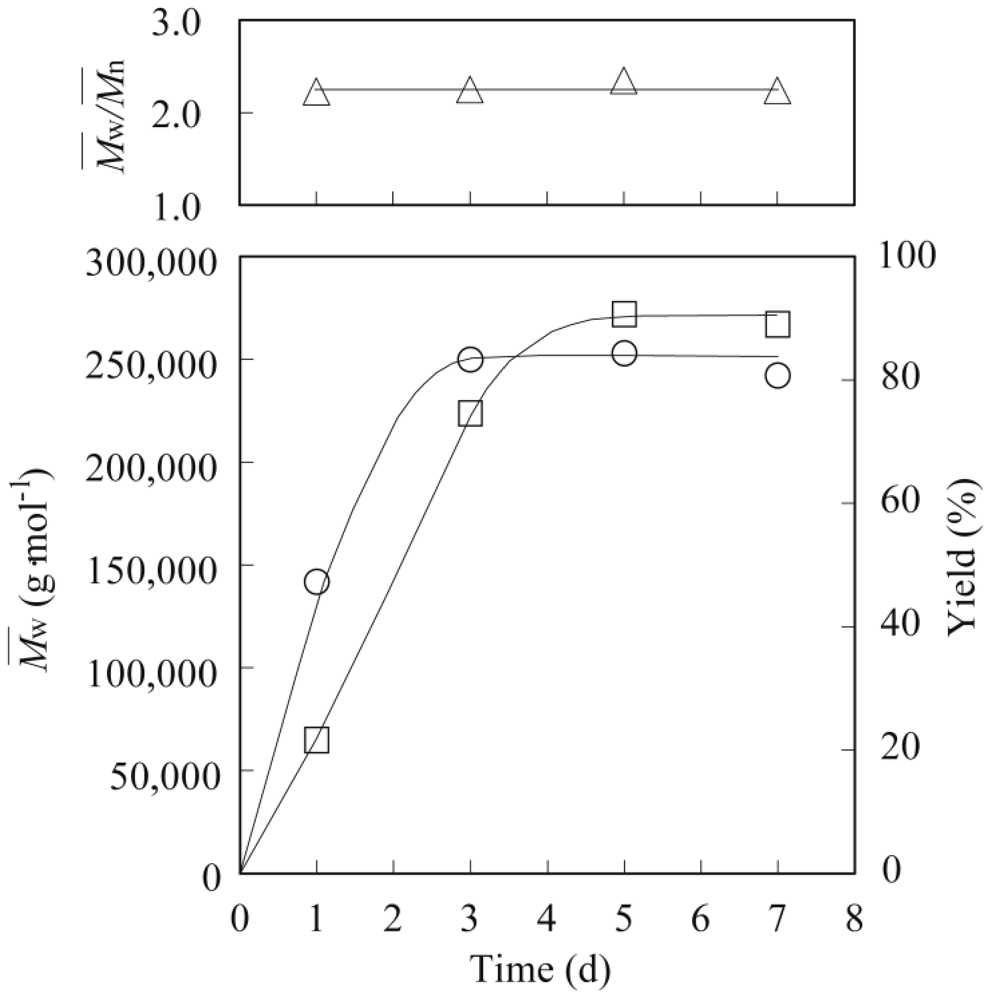
3.2. Lipase-Catalyzed Polymerization of Methyl Epoxyricinoleate

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Lipase a) | Mw (g mol−1) | Mw/Mn |
|---|---|---|---|
| 1 | Candida antarctica (CALB) | 68,700 | 2.2 |
| 2 | Candida rugosa (lipase CR) | <800 | 1.1 |
| 3 | Burkholderia cepacia (lipase PS-IM) | 99,100 | 2.4 |
| 4 | Pseudomonas fluorescens (lipase AK) | 24,800 | 1.8 |
| 5 | Thermally diactivated lipase PS-IM | <800 | 1.1 |
| 6 | Blank | <800 | 1.1 |
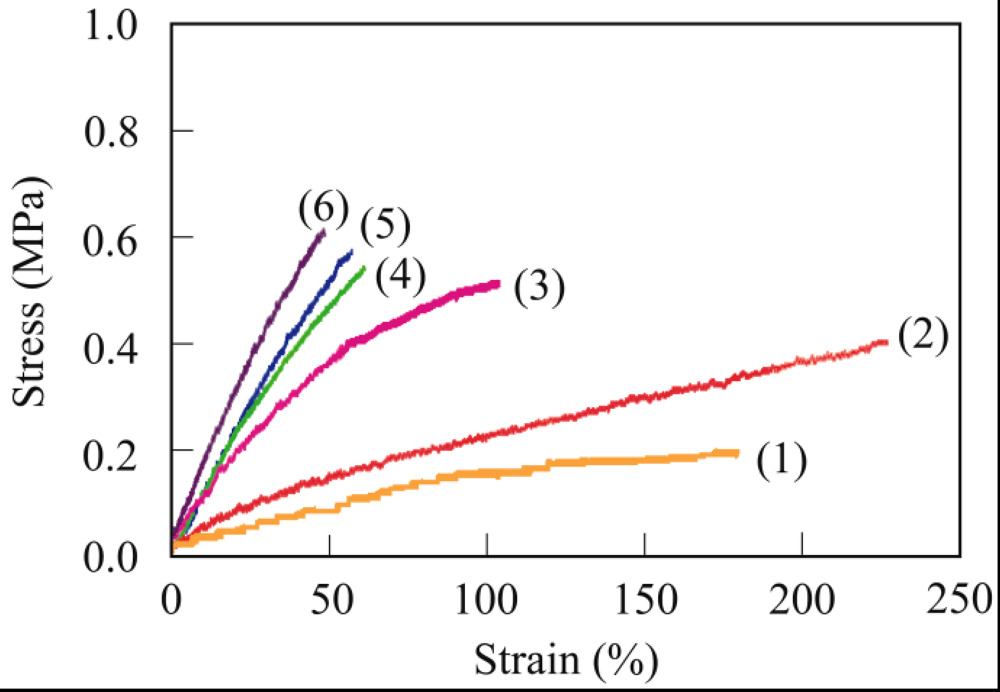
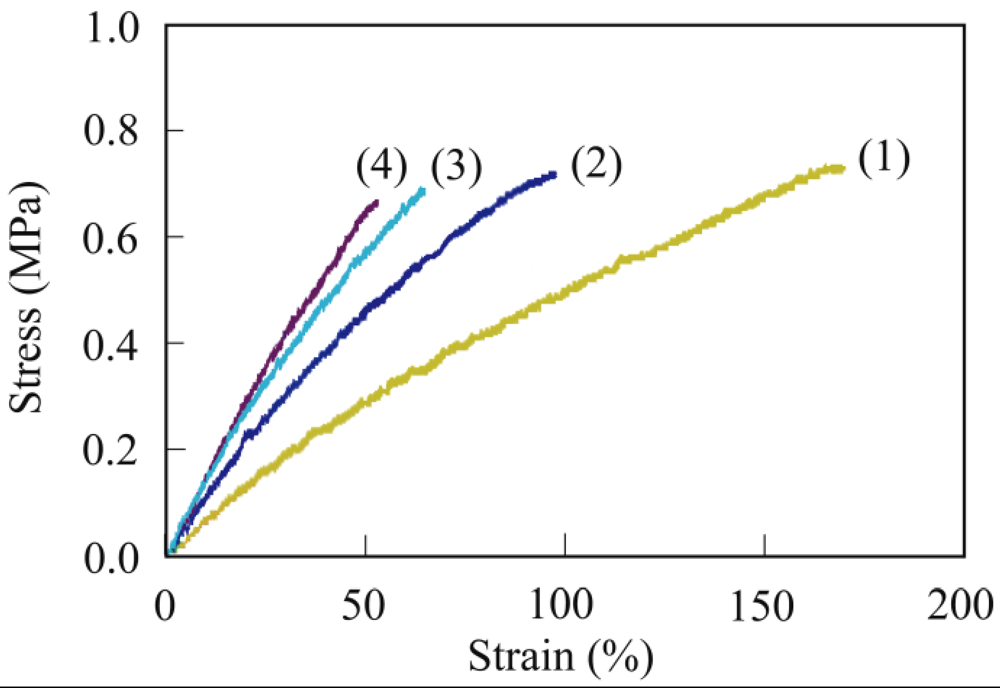
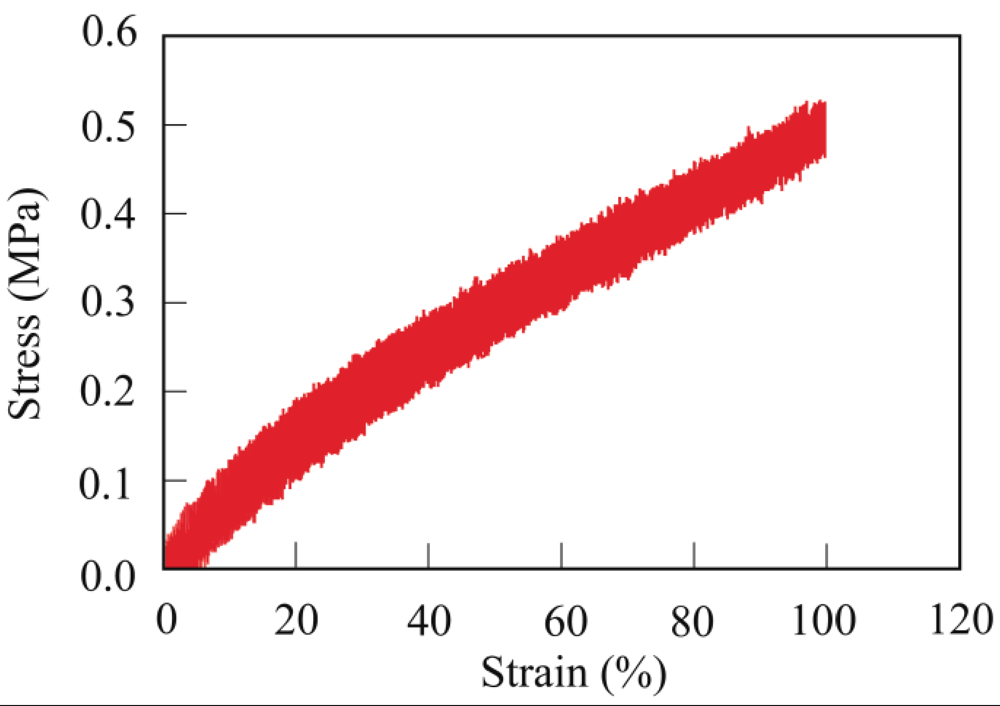
3.3. Crosslinking of PER
3.4. Thermal Properties
| Crosslinker Content (mol%) | Mw of PER (g·mol−1) | Tg (°C) | Young’s Modulus (MPa) | Tensile Strength (MPa) | Elongation at Break (%) | G' (25 °C) (MPa) | G' (Tg +50 °C) (MPa) | |
|---|---|---|---|---|---|---|---|---|
| PER-MA | 20 | 186,000 | −17.8 a) | 0.408 | 0.404 | 220 | - | - |
| 40 | 186,000 | −12.4 a) | 0.850 | 0.507 | 101 | - | - | |
| 60 | 186,000 | −8.8 a) | 1.11 | 0.543 | 60.1 | - | - | |
| 80 | 186,000 | −2.9 a) | 1.25 | 0.573 | 56.4 | - | - | |
| 100 | 186,000 | 2.2 a) | 1.38 | 0.621 | 48.8 | - | - | |
| 100 | 94,000 | −5.5 b) | 0.638 | 0.715 | 171 | 0.890 | 0.744 | |
| 100 | 147,000 | 5.1 b) | 1.08 | 0.680 | 102 | 3.89 | 1.24 | |
| 100 | 172,000 | 15.0 b) | 1.30 | 0.693 | 64.0 | 5.98 | 2.10 | |
| 100 | 224,000 | 22.0 b) | 1.49 | 0.675 | 47.7 | 13.8 | 4.76 | |
| PER-SA | 100 | 186,000 | −4.2 a) | 0.156 | 0.220 | 178 | - | - |
3.4. Mechanical Properties
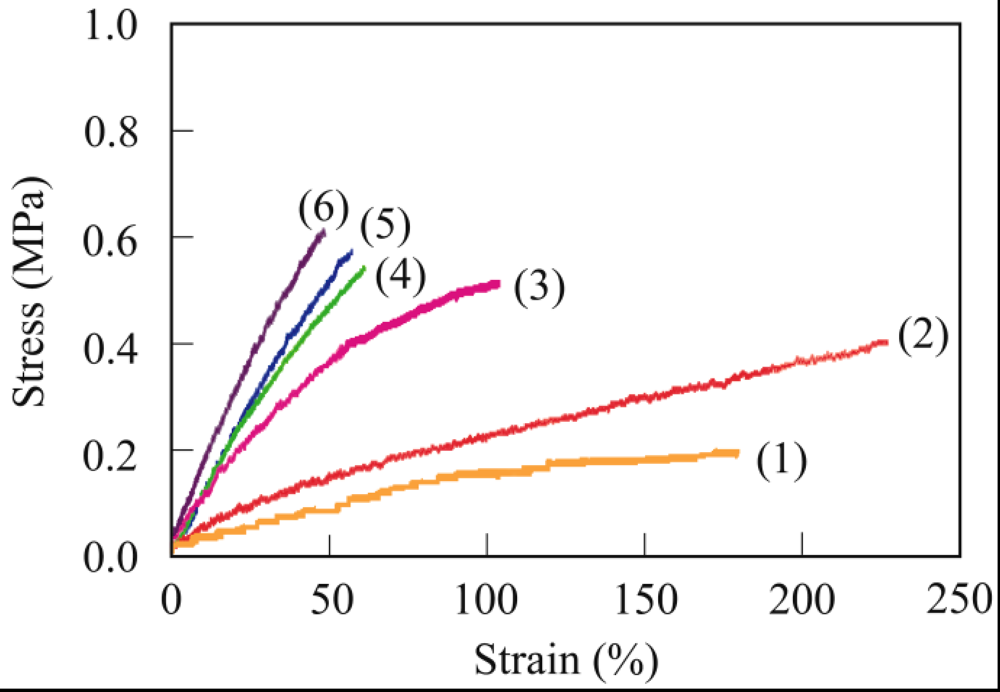
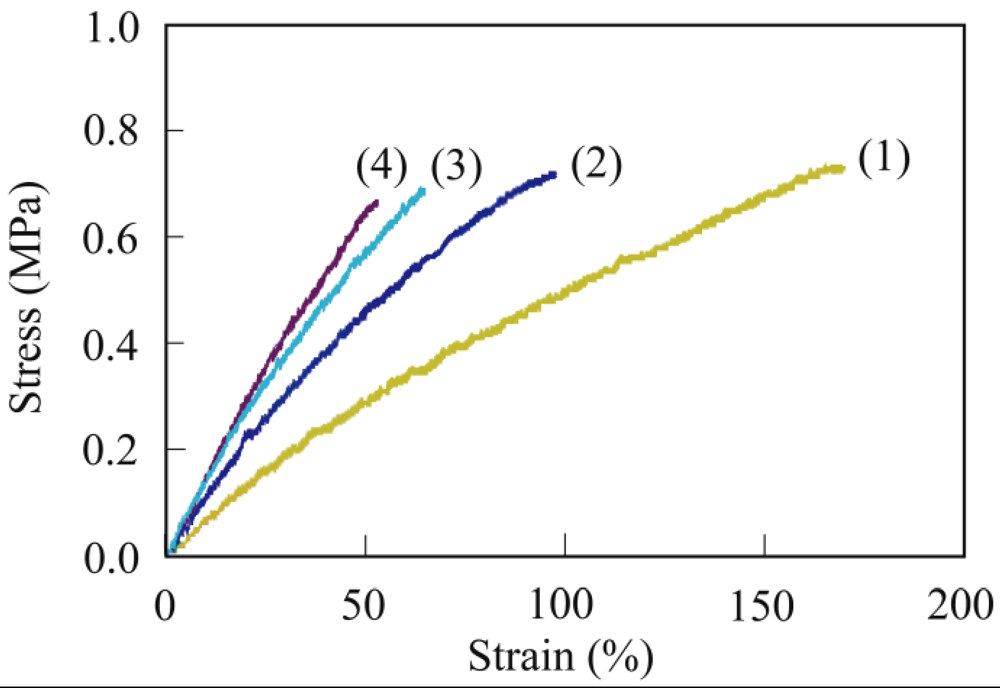
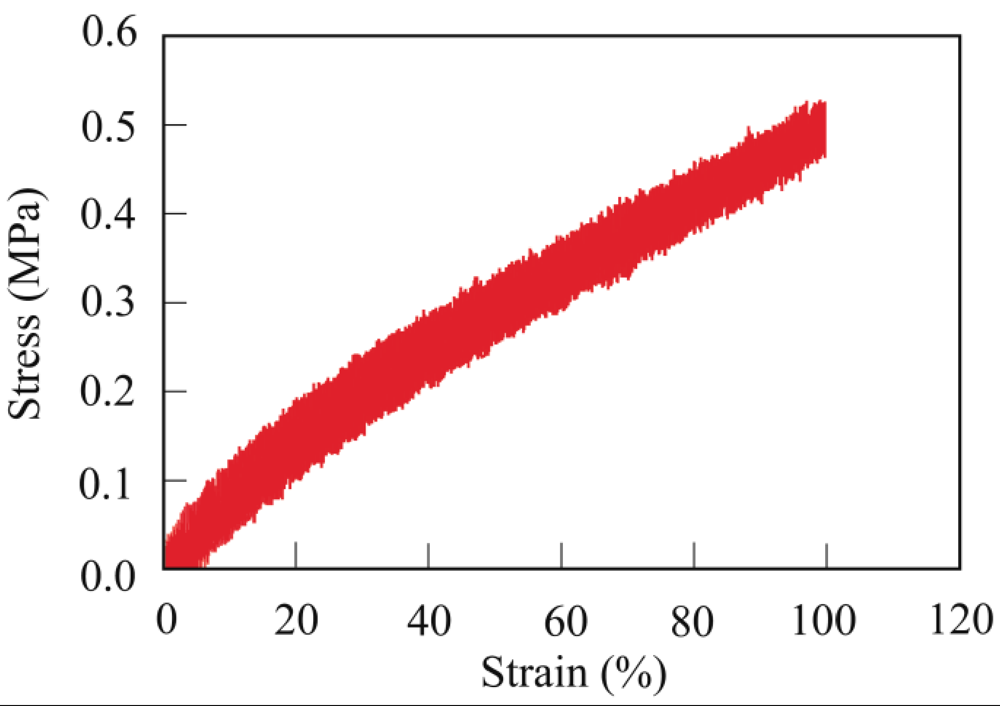
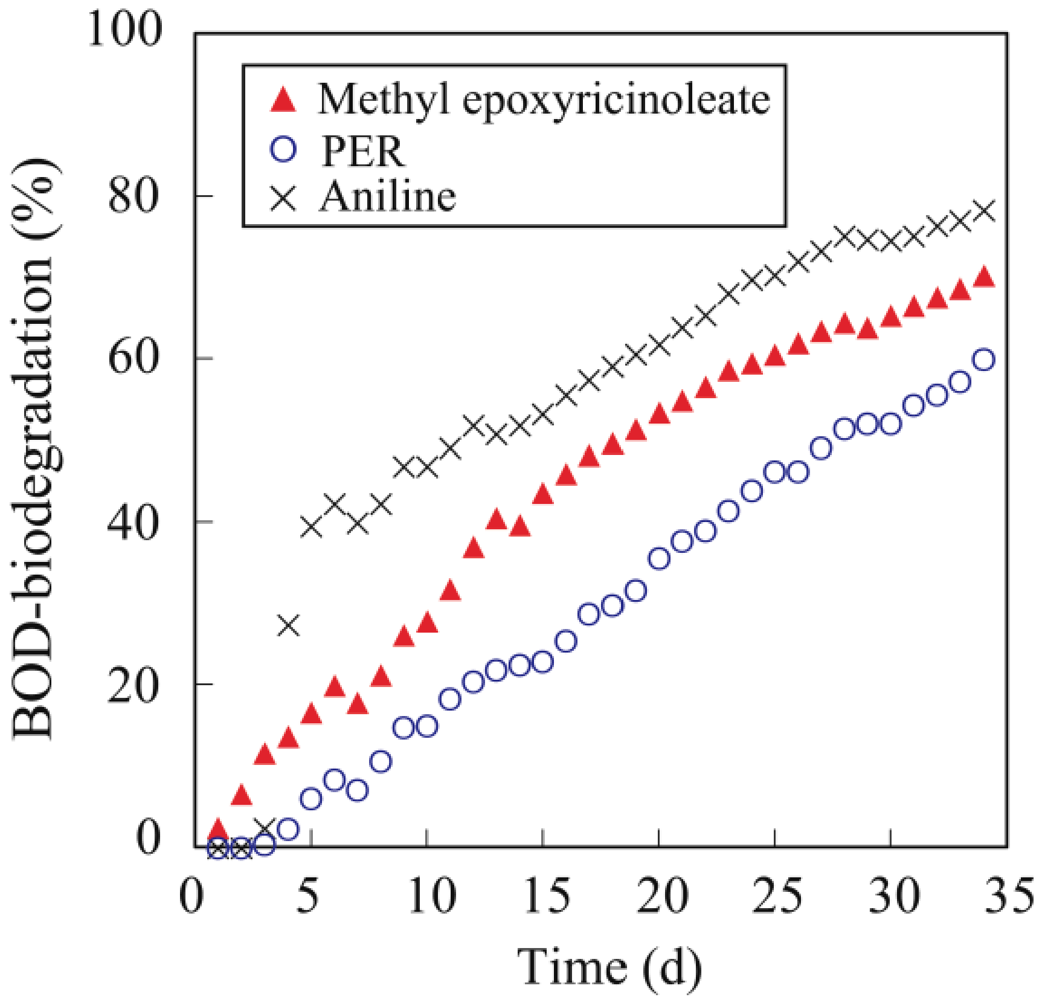
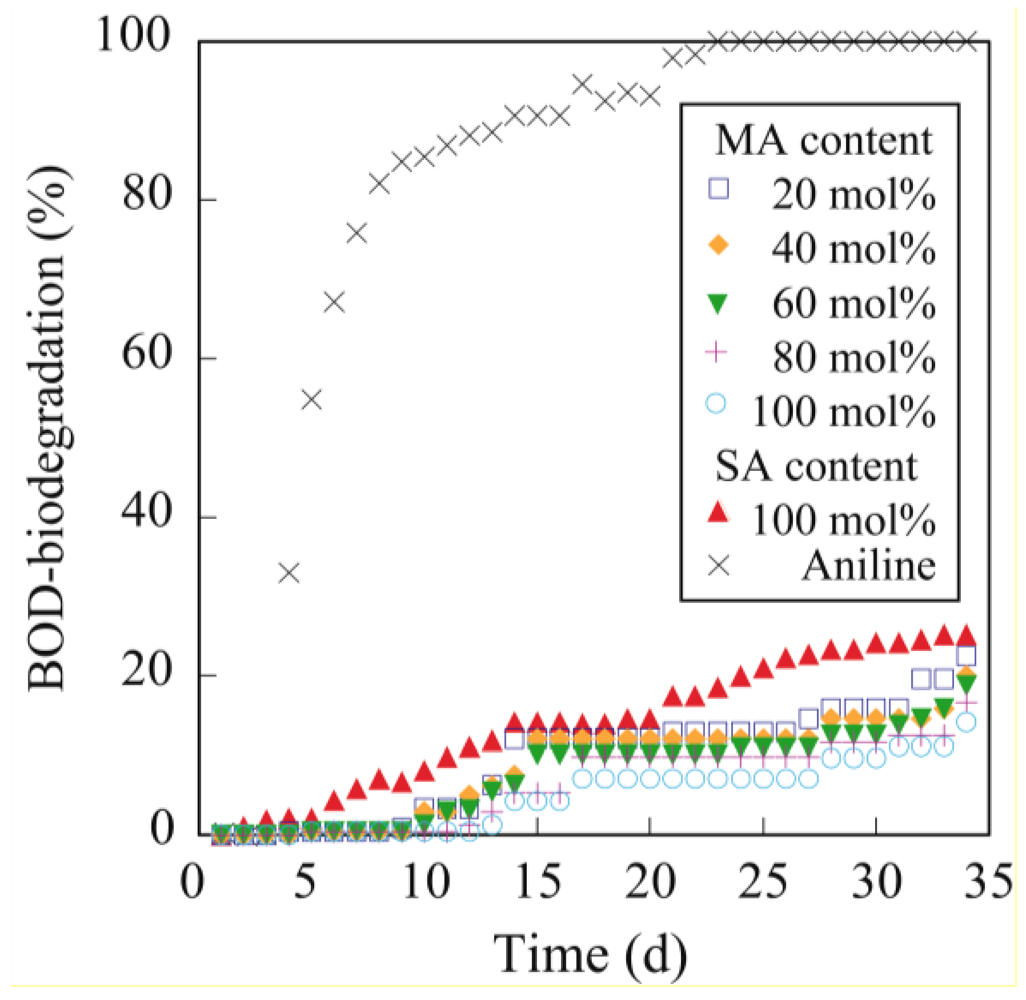
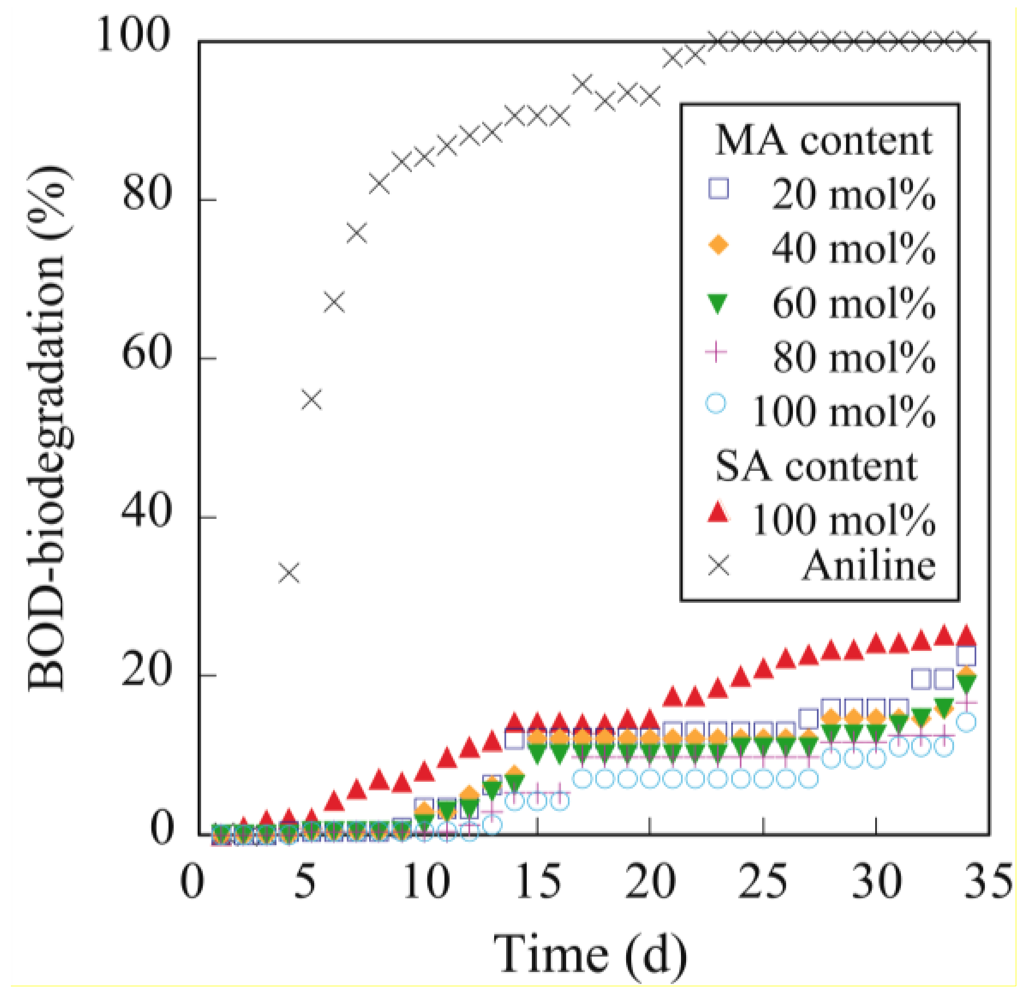
3.5. Biodegradability
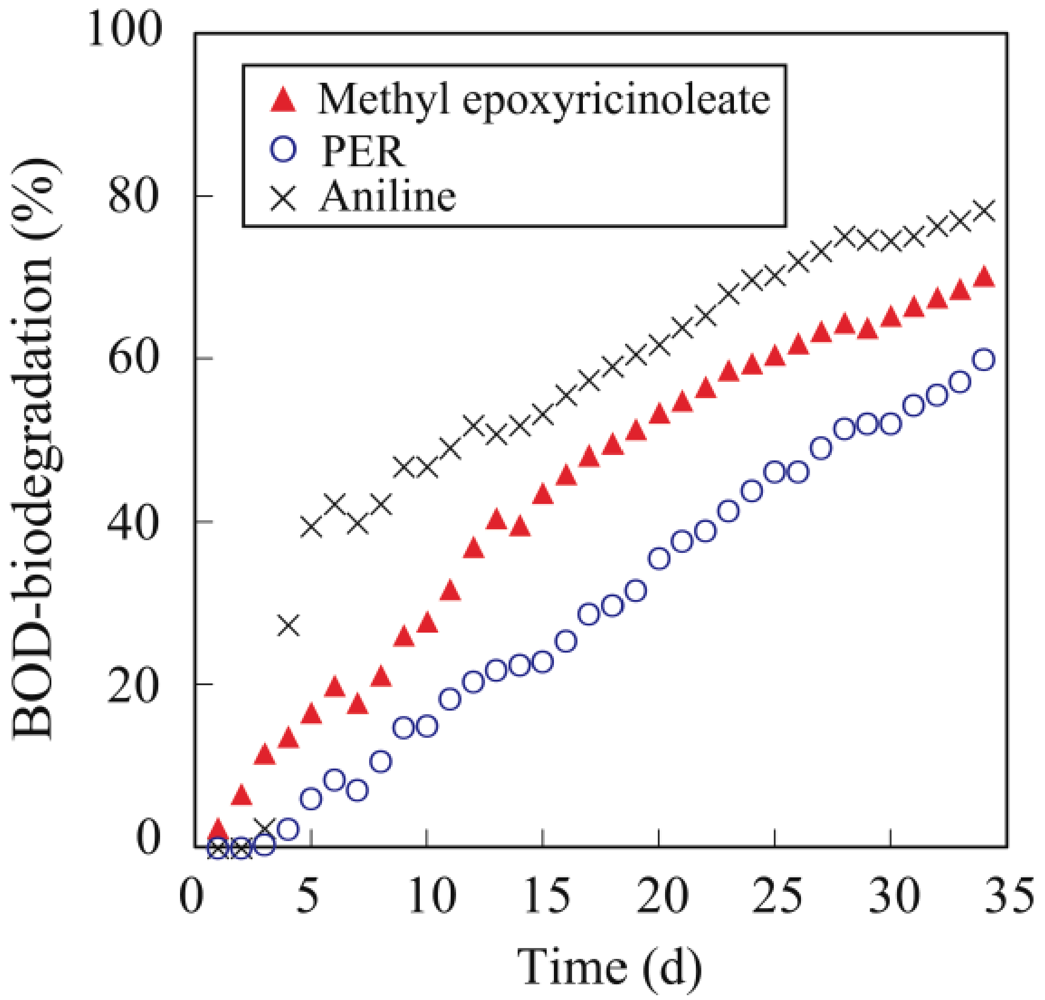
4. Conclusions
Acknowledgments
References
- Pepic, D.; Nikolic, M.S.; Djonlagic, J. Synthesis and characterization of biodegradable aliphatic copolyesters with poly(tetramethylene oxide) soft segments. J. Appl. Polym. Sci. 2007, 106, 1777–1786. [Google Scholar]
- Alexander, H. Growing plastics. Chem. Eng. News 2008, 86, 21-22, 24-25. [Google Scholar]
- Yasuda, M.; Ebata, H.; Matsumura, S. Enzymatic synthesis and properties of novel biobased elastomers consisting of 12-hydroxystearate, itaconate and butane-1,4-diol. ACS Symp. Ser. 2010, 1043, 237–251. [Google Scholar]
- Biermann, U.; Friedt, W.; Lang, S.; Luhs, W.; Machmuller, G.; Metzger, J.O.; Klass, M.R.; Schafer, H.J.; Schneider, M.P. New synthesis with oils and fats as renewable raw materials for the chemical industry. Angew. Chem. Int. Ed. 2000, 39, 2206–2224. [Google Scholar]
- Tsujimoto, T.; Uyama, H.; Kobayashi, S. Enzymatic synthesis and curing of biodegradable crosslinkable polyesters. Macromol. Biosci. 2002, 7, 329–335. [Google Scholar]
- Ebata, H.; Yasuda, M.; Toshima, K.; Matsumura, S. Poly (Ricinoleic Acid) based novel thermosetting elastomer. J. Oleo Sci. 2008, 57, 315–320. [Google Scholar]
- Ebata, H.; Toshima, K.; Matsumura, S. Lipase-catalyzed synthesis and cuing of high-molecular-weight polyricinoleate. Macromol. Biosci. 2007, 7, 798–803. [Google Scholar]
- Kobayashi, T.; Matsumura, S. Enzymatic synthesis and properties of novel biodegradable and biobased thermoplastic elastomers. Polym. Degrad. Stab. 2011, 96, 2071–2079. [Google Scholar]
- Ozturk, C.; Kusefoglu, S.H. Polymerization of epoxidized soybean oil with maleinized soybean oil and maleic anhydride grafted polypropylene. J. Appl. Polym. Sci. 2010, 118, 3311–3317. [Google Scholar]
- Lukaszczyk, J.; Jaszcz, K. Studies on hydrolytic degradation of epoxy-polyester resins cured with glutaric anhydride. Polym. Adv. Technol. 2002, 13, 871–883. [Google Scholar]
- Vlcek, T.; Petrovic, Z.S. Optimization of the chemoenzymatic epoxidation of soybean oil. J. Am. Oil Chem. Soc. 2006, 83, 247–252. [Google Scholar]
- Miao, S.; Zhang, S.; Su, Z.; Wang, P. Chemoenzymatic synthesis of oleic acid-based polyesters for use as highly stable biomaterials. J. Polym. Sci. 2008, 46, 4243–4248. [Google Scholar]
- Modified MITI Test, OECD Guidelines for Testing of Chemicals; Organization for Economic Cooperation and Development (OECD): Paris, France, 1981; p. 301C.
- Matsumura, S.; Ebata, H.; Toshima, K. A new strategy for sustainable polymer recycling using an enzyme: Poly(e-caprolactone). Macromol. Rapid Commun. 2000, 21, 860–863. [Google Scholar]
- Ebata, H.; Toshima, K.; Matsumura, S. A strategy for increasing molecular weight of polyester by lipase-catalyzed polymerization. Chem. Lett. 2001, 8, 798–799. [Google Scholar]
- Kato, M.; Toshima, K.; Matsumura, S. Preparation of aliphatic poly(thioester) by the lipase-catalyzed direct polycondensation of 11-mercaptoundecanoic acid. Biomacromolecules 2005, 6, 2275–2280. [Google Scholar]
- Sugihara, S.; Toshima, K; Matsumura, S. New strategy for enzymatic synthesis of high-molecular-weight poly(butylene succinate) via cyclic oligomers. Macromol. Rapid Commun. 2006, 27, 203–207. [Google Scholar] [CrossRef]
- Bjorkling, F.; Godtfredsen, S.E.; Kirk, O. Lipase-mediated formation of peroxycarboxylic acids used in catalytic epoxidation of alkenes. J. Chem. Soc. Chem. Commun. 1990, 1301–1303. [Google Scholar]
- Warwel, S.; Klaas, M.R.G. Chemo-enzymatic epoxidation of unsaturated carboxylic acids. J. Mol. Catal. B Enzym. 1995, 1, 29–35. [Google Scholar]
- Svedendahl, M.; Carlqvist, P.; Branneby, C.; Allner, O.; Frise, A.; Hult, K.; Berglund, P.; Brinck, T. Direct epoxidation in Candida antarctica lipase B studied by experiment and theory. ChemBioChem 2008, 9, 2443–2551. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kazariya, A.; Matsumura, S. Enzymatic Synthesis and Crosslinking of Novel High Molecular Weight Polyepoxyricinoleate. Polymers 2012, 4, 486-500. https://doi.org/10.3390/polym4010486
Kazariya A, Matsumura S. Enzymatic Synthesis and Crosslinking of Novel High Molecular Weight Polyepoxyricinoleate. Polymers. 2012; 4(1):486-500. https://doi.org/10.3390/polym4010486
Chicago/Turabian StyleKazariya, Ayaki, and Shuichi Matsumura. 2012. "Enzymatic Synthesis and Crosslinking of Novel High Molecular Weight Polyepoxyricinoleate" Polymers 4, no. 1: 486-500. https://doi.org/10.3390/polym4010486
APA StyleKazariya, A., & Matsumura, S. (2012). Enzymatic Synthesis and Crosslinking of Novel High Molecular Weight Polyepoxyricinoleate. Polymers, 4(1), 486-500. https://doi.org/10.3390/polym4010486