1. Introduction
Polymeric nanocarriers have gained significant attention for biomedical applications owing to their structural tunability, pH-responsiveness, and ability to form stable nanostructures [
1,
2]. Among these, amphiphilic block copolymers are particularly advantageous, as they self-assemble into well-defined nanoscale structures such as micelles. These micellar systems feature a hydrophilic outer shell, which enhances stability in physiological environments, and a hydrophobic core, which can host hydrophobic guest molecules or functional moieties within the core [
3].
Poly(ethylene glycol) (PEG) is widely used in biomedical formulations due to its hydrophilicity, low immunogenicity, and proven safety profile [
4,
5,
6,
7]. The incorporation of PEG in polymeric nanocarriers reduces protein adsorption, enhances colloidal stability, and prevents rapid clearance by the immune system, thereby influencing their biodistribu istion and interaction with biological systems [
6,
7]. In addition, the size and molecular composition of nanocarriers significantly influence their pharmacokinetics and biodistribution. Nanocarriers in the 10–200 nm range demonstrate optimal circulation behavior, avoiding rapid renal clearance while taking advantage of the enhanced permeability and r is notetention (EPR) effect for passive targeting [
8,
9].
The incorporation of imidazole moieties into polymeric structures has also attracted interest in biomedical applications due to their distinct physicochemical properties. Imidazole, a nitrogen-containing heterocyclic ring, plays a key role in biological interactions, including enzymatic catalysis and molecular recognition [
10]. Synthetic polymers bearing imidazole groups have been investigated for drug delivery applications, as they exhibit hydrogen bonding capabilities, pH responsiveness, and metal coordination properties, which make them attractive candidates for stimuli-responsive and functional polymer architectures [
11,
12,
13,
14,
15]. Various polymerization techniques, including free radical polymerization, nitroxide-mediated polymerization (NMP), and group transfer polymerization (GTP) [
13], have been explored for the synthesis of imidazole-functionalized polymers. However, reversible addition–fragmentation chain transfer (RAFT) polymerization [
16] has emerged as a preferred approach due to its ability to precisely control molecular weight and copolymer composition, enabling the fabrication of well-defined amphiphilic materials [
15,
17,
18,
19].
However, to the best of our knowledge, PEG-1-vinyl imidazole (PEG-
b-VIM) diblock copolymers have not been previously reported, nor has their use been explored in the context of radiolabeling. The synthesis of such amphiphilic copolymers is challenging due to the polarity and coordinating nature of the imidazole monomer, which can interfere with controlled polymerization [
12]. Furthermore, while imidazole-based copolymers have been explored for biomedical use, their potential in radiometal chelation and in vivo tracking remains underexplored.
In this study, we report the first example of PEG-b-1-vinyl imidazole (VIM) diblock copolymers synthesized using RAFT polymerization. The resulting copolymers were characterized by 1H NMR spectroscopy to confirm their molecular composition, and their polymerization behavior was systematically evaluated. Furthermore, a post-polymerization modification via aminolysis enabled the conjugation of a NOTA chelator to study their pharmacokinetics.
2. Experimental Section
Materials and Methods. All chemical reagents used in this study were of analytical grade and obtained from Sigma-Aldrich Germany, Acros Organics Leicestershire, UK, and Fluka, Roumania. Unless otherwise stated, they were used as received without further purification. High-performance liquid chromatography (HPLC)-grade solvents were used for all analytical procedures and degassed using a continuous helium flux. Deuterated solvents, including deuterated chloroform (CDCl3, 99.8%), deuterated dimethyl sulfoxide (d6-DMSO, 99.9%), and deuterium oxide (D2O), were purchased from Merck, Germany. Tetrahydrofuran (THF, 99.8%) was sourced from Labscan Analytical Science Gliwice, Poland. For the synthesis of the Macro-DMPA chain transfer agent, poly(ethylene oxide) (PEO) with molecular weights Mn = 750 g/mol, Mn = 2000 g/mol, and Mn = 5000 g/mol, 4-dimethylaminopyridine (DMAP), dicyclohexylcarbodiimide (DCC), 2-Dodecylthiocarbonylthio-2-methylpropanoic acid (DMPA), tripotassium phosphate, and carbon disulfide were used along with 1-dodecanethiol, all obtained from Sigma-Aldrich, Germany. The polymerization of VIM was carried out using reversible addition–fragmentation chain transfer (RAFT) polymerization, with 2,2′-Azobis(2-methylpropionitrile) (AIBN, 98%) as the radical initiator and glacial acetic acid (100%) as the reaction medium. 1-Vinyl Imidazole (VIM, >99%) was purified by passage through a basic alumina column, followed by stirring over calcium hydride to eliminate residual moisture and protic contaminants. A small quantity of 2,2-diphenyl-1-picrylhydrazyl hydrate (DPPH, 95%) was added as a radical inhibitor. The purified VIM was stored at 5 °C and vacuum-distilled immediately prior to use to ensure high purity.
To improve the biomedical applicability of the copolymers, post-polymerization modification was performed via aminolysis. Diazabicyclo [5.4.0]undec-7-ene (DBU) was used to remove the trithiocarbonate end-group, facilitating the conjugation of a NOTA chelator (2,2′-(7-(2-((2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethyl)amino)-2-oxoethyl)-1,4,7-triazonane-1,4-diyl)diacetic acid, Maleimide-NOTA, sourced from Chematech, Dijon France) for radiolabeling.
For polymer radiolabeling, sodium pertechnetate (Na
99mTcO
4) was obtained in physiological saline from a commercial
99Mo/
99mTc generator (Ultra-Technekow™ V4 Generator, Curium Pharma, Petten, The Netherlands). The precursor
fac-[
99mTc(CO)
3(H
2O)
3]
+ was synthesized in-house using a reaction kit containing 5.5 mg NaBH
4, 4 mg Na
2CO
3, and 20 mg Na-K tartrate. After purging with carbon monoxide gas, Na
99mTcO
4 was introduced following established literature procedures to obtain the final radiolabeled species [
20].
All chromatographic analyses were performed using a Waters 600 system (Waters, Belgium) equipped with a Waters 2487 Dual λ absorbance detector and a Gabi gamma detector (Raytest, Germany). Separations were conducted on a Macherey-Nagel Nucleosil RP-C18 column (10 μm, 250 × 4 mm) using a binary gradient system. The mobile phase consisted of water with 0.1% trifluoroacetic acid (TFA) (Phase A) and methanol with 0.1% TFA (Phase B). The gradient began at 90% A (10% B) for 1 min, then linearly shifted to 10% A (90% B) over the next 9 min, maintaining this composition for an additional 10 min.
Synthesis of 2-Dodecylthiocarbonylthio-2-methylpropanoic Acid (DMPA). The chain transfer agent was synthesized following established protocols [
21]. In a 250 mL round-bottom flask, 8.39 g of tripotassium phosphate (39.52 mmol), 9.5 mL of 1-dodecanethiol (8 g, 39.52 mmol), and 50 mL of acetone were mixed and stirred for 30 min. Following this, 6.5 mL of carbon disulfide (107.79 mmol) was added, leading to a visible yellow coloration of the solution. After an additional 30 min, 6 g of 2-bromo-2-methylpropionic acid (35.93 mmol) was introduced, and the reaction was maintained under stirring for 24 h. The reaction mixture was then filtered, acidified using 1 M HCl, and extracted with DCM. The organic layers were dried over anhydrous magnesium sulfate, and the crude product was purified via column chromatography using a hexane/ethyl acetate (90:10) mobile phase. The final product, obtained as a yellow solid in 53.4% yield, was structurally confirmed using
1H-NMR spectroscopy.
Synthesis of Macro PEG-DMPA. Three variations of Macro-DMPA were prepared via esterification of poly(ethylene glycol) methyl ether (m-PEG-OH) with different molecular weights (750 g/mol (DP = 16), 2000 g/mol (DP = 45), and 5000 g/mol (DP = 114)). For the synthesis of m-PEG45-DMPA, a mixture of 3 g of m-PEG45 (1.5 mmol), 0.82 g of DMPA (2.25 mmol), and 0.02745 g of DMAP (0.22 mmol) was dissolved in 10 mL of anhydrous DCM within a 50 mL round-bottom flask. The system was sealed and purged with argon to eliminate residual oxygen. Subsequently, 0.4635 g of DCC (2.25 mmol), dissolved in 5 mL of dry DCM, was added dropwise. The reaction was conducted under constant stirring at room temperature for 24 h. The crude reaction mixture was filtered to remove dicyclohexylurea byproducts, concentrated using a rotary evaporator, and further purified by silica column chromatography using a DCM/methanol (95:5) eluent system. The purity of the final product was confirmed through thin-layer chromatography (TLC) and 1H-NMR spectroscopy.
Synthesis of Diblock Copolymers PEGx-b-PVIMy. All diblock copolymers were synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization. Before polymerization, vinyl imidazole (VIM) was purified using a basic alumina column to remove acidic impurities. A drop of 2,2-diphenyl-1-picrylhydrazyl (DPPH) was added to inhibit radical side reactions. A representative synthesis of PEG45-b-PVIM100 was carried out in a 50 mL Schlenk flask containing 0.6 g (6 mmol) of macro-PEG45-DMPA, 0.192 mL of VIM, 6.2 mg of AIBN (0.04 mmol), and 6.1 mL of acetic acid. The solution was degassed using three freeze-pump-thaw cycles and then heated at 70 °C for 22 h. The final product was precipitated in ether, collected, dried under vacuum, and stored.
Conjugation of Diblock Copolymers PEG-b-PVIM with Chelating Agent NOTA. A 50 mL Schlenk flask was charged with 0.2 g of PEG45-b-PVIM100 (2.265 × 10−5 mol) and an appropriate amount of maleimide-NOTA (2.265 × 10−6 mol, 0.1 equivalents), dissolved in dry DMF. Separately, DBU was dissolved in DMF and added to the reaction mixture. The reaction proceeded at 70 °C for 24 h. The final product was purified via recrystallization in ether, collected, dried under vacuum, and stored.
Synthesis of 99mTc Complexes. The
fac-[
99mTc(CO)
3(H
2O)
3]
+ precursor was prepared using the homemade kit, and its radiochemical purity was confirmed by reverse-phase HPLC. A solution (0.5–1.0 mL, 37–740 MBq, pH 6) of this precursor was mixed in a capped vial with 100 μg of each macro-compound [Macro-PEG
16(
99mTc-1), PEG
16-
b-PVIM
48(
99mTc-2), PEG
114-
b-PVIM
86(
99mTc-3), PEG
114-
b-PVIM
48(
99mTc-4), PEG
45-
b-PVIM
34(
99mTc-5), PEG
45-
b-PVIM
69(
99mTc-6), and Macro-PEG
45(
99mTc-7)]. The mixture was incubated at 80 °C for 30 min, followed by HPLC analysis [
22]. For subsequent stability and animal studies, each complex was purified by HPLC to achieve a radiochemical purity (RCP) of 99.5%. Solvents were then removed under a gentle flow of N
2 at 40 °C, and the radiotracers were reconstituted in phosphate-buffered saline (PBS, 10 mM, pH 7.4) containing 10% ethanol.
In Vitro Stability Studies of 99mTc Complexes. HPLC-purified samples of the
99mTc complexes were kept at room temperature for up to 6 h. To assess stability, challenge experiments were conducted by adding 0.8 mL of PBS (pH 7.4) and 0.1 mL of a 0.1 M aqueous solution of either cysteine or histidine to 0.1 mL (3.7 MBq) of each
99mTc complex, followed by incubation at 37 °C. Samples were collected at 1, 3, and 6 h for HPLC analysis. For serum stability tests, 0.1 mL of each tracer (3.7 MBq) was mixed with 0.4 mL of human serum and incubated at 37 °C. Non-specific serum protein binding was determined by precipitating proteins with 0.5 mL of acetonitrile, centrifuging at 5000 rpm for 5 min, and collecting the supernatant. The protein pellet was redissolved in 0.2 mL acetonitrile and centrifuged again. The combined supernatants were measured using a dose calibrator to calculate the binding percentage. Subsequently, the acetonitrile was removed by gentle heating at 40 °C under a stream of N
2, and the residues were reconstituted in saline (with 10% ethanol) before analysis by radio-HPLC [
23].
Polymer Characterization. Gel Permeation Chromatography (GPC). The molecular weights (MW) and molecular weight distributions (MWDs) of the synthesized copolymers were determined using GPC. A Waters 2695 system equipped with a Waters 2414 refractive index detector (Waters, Belgium) and an Ultrahydrogel linear column (10 μm, 7.8 mm × 300 mm, 1K–7M) was used. The mobile phase consisted of 0.1 M NaNO3 in H2O at a flow rate of 1 mL/min. Calibration was performed using nine narrow-distribution PEG standards (106, 390, 1010, 1490, 4070, 10,300, 19,500, 31,700, and 72,300 g/mol). The number-average molecular weight (Mn), weight-average molecular weight (Mw), and polydispersity index (Mw/Mn) were calculated.
Nuclear Magnetic Resonance Spectroscopy (NMR). 1H-NMR spectra of DMPA, MacroPEG-DMPA, PEGx-b-PVIMy copolymers, and PEGx-b-PVIMy-NOTA conjugates were recorded in D2O, CDCl3, and d6-DMSO using Bruker Avance 300 and 500 MHz spectrometers to confirm the structure and composition.
Hydrogen ion titration. The protonation behavior of the diblock copolymers was investigated through acid–base titration. Aqueous solutions (0.01 wt%) of each copolymer were titrated with a standard 1 M NaOH solution from pH 2 to 11 and monitored using a HANNA HI991300 portable pH meter. Furthermore, the effective pK of the PVIM units was estimated as the pH at 50% ionization.
Dynamic Light Scattering (DLS). The hydrodynamic diameters of the synthesized diblock copolymers in H2O were measured using a Brookhaven 90Plus DLS spectrophotometer with a BI9000 correlator and a 30 mW red diode laser (673 nm) at a 90° angle. A 1 wt% polymer solution was filtered through a 0.45 μm PTFE syringe filter, allowed to settle for one hour to remove air bubbles, and analyzed using multimodal size distribution (MSD) analysis based on non-negatively constrained least squares (NNCLS).
Distribution Coefficient (D
o/w) Determination. The lipophilicity of the
99mTc complexes was determined using the shake-flask method. In brief, 10 μL (approximately 370 kBq) of the purified complex was added to a centrifuge tube containing 3 mL of a 1:1 mixture of 1-octanol and PBS (0.1 M, pH 7.4). After vortexing for 1 min at room temperature, the mixture was centrifuged at 5000 rpm for 5 min. Radioactivity from three separate 0.1 mL aliquots from both the 1-octanol and PBS layers was measured using a gamma counter. Additional partitioning of a 0.5 mL aliquot of the octanol phase was performed until consistent counts were obtained. This procedure was repeated three times, and the D
o/w was calculated as the ratio of counts in the octanol phase to those in the PBS phase [
23].
Imaging Studies/Imaging System. Real-time dynamic imaging was performed using a specialized, mouse-sized planar scintigraphy system (γ-eye™ by BIOEMTECH, Athens, Greece). This system fuses scintigraphic data with digital photographs of the subject and employs a deep neural network to generate synthetic X-ray images for enhanced anatomical co-registration [
24]. Based on position-sensitive photomultiplier tubes (PSPMTs) coupled with a CsI(Na) pixelated scintillator and a medium-energy lead collimator with parallel hexagonal holes, the detector is optimized for various SPECT isotopes. The system is well-suited for high-quality planar imaging with a field of view of 5 × 10 cm
2 and a spatial resolution of approximately 2 mm. During imaging sessions, healthy Swiss Albino mice were maintained under isoflurane anesthesia and at a constant temperature of 37 °C. Short static scans (typically 10 min or less) were acquired at multiple time points to monitor tracer distribution over time [
25]. Image post-processing and quantification were performed using the embedded visual|eyes software (BIOEMTECH, Athens, Greece).
Animal Imaging Studies. For SPECT imaging, mice received intravenous bolus injections of
99mTc tracers (0.1 mL, ~10 MBq) under isoflurane anesthesia (induction at 3–5% and maintenance at 1–3%). Imaging was conducted on live animals to capture real-time biodistribution data [
26]. All the biodistribution experiments were carried out in compliance with the national laws and European protocols (2010/63/EU) related to the conduct of animal experimentation. The corresponding animal experiments were approved by the Hellenic Ministry of Rural Development and Food (426573/03-04-2024).
3. Results and Discussion
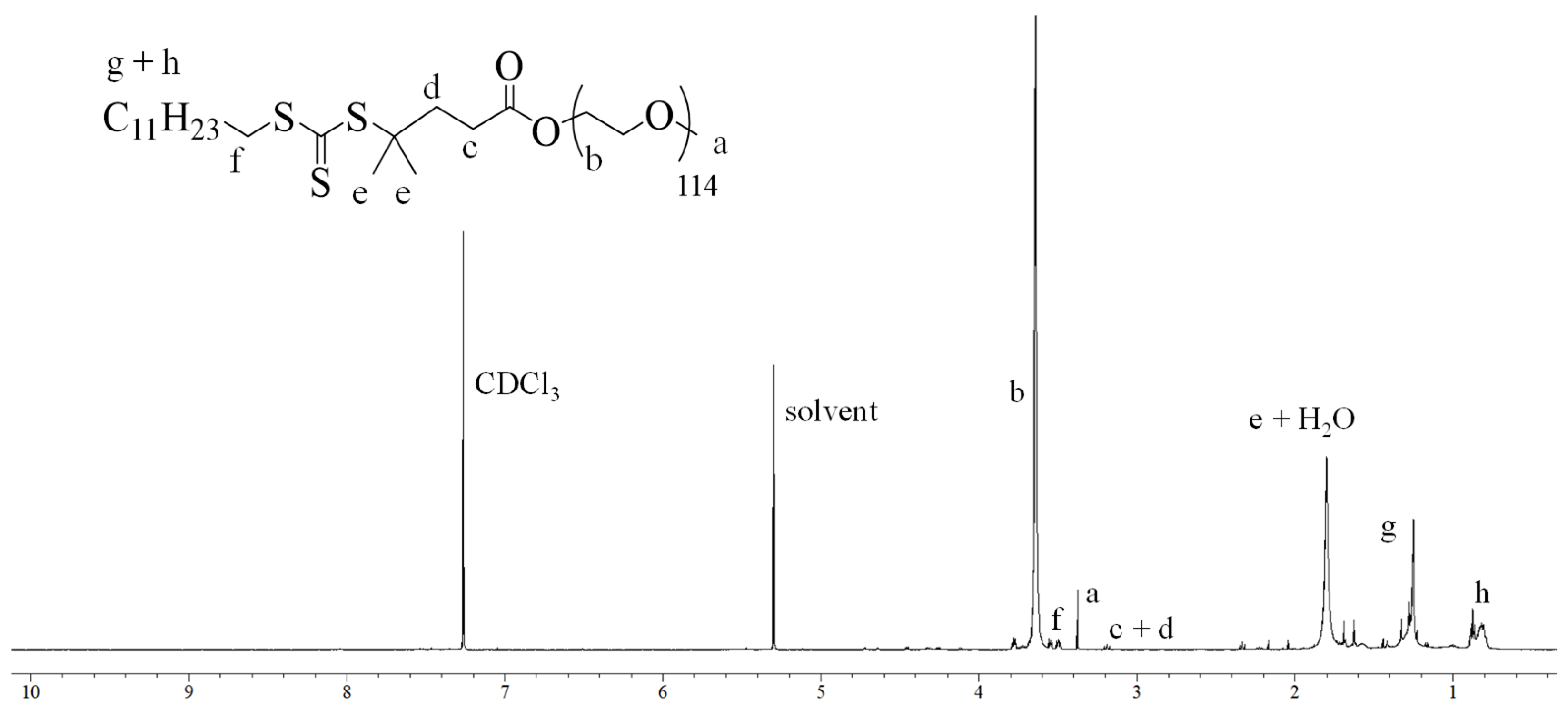
Synthesis and Characterization of Macro-PEG-DMPA. The synthesis of the macro chain transfer agent (MacroPEG-DMPA) was carried out via Steglich esterification, a mild and efficient method that ensures high conversion rates while maintaining the integrity of sterically demanding substrates. The reaction involved the coupling of poly(ethylene glycol) methyl ether (m-PEG-OH) with 2-Dodecylthiocarbonylthio-2-methylpropanoic acid (DMPA) in the presence of dicyclohexylcarbodiimide (DCC) as the coupling agent and 4-dimethylaminopyridine (DMAP) as the catalyst. This reaction sequence resulted in the successful formation of Macro-PEG-DMPA, which was subsequently purified through multiple steps, including filtration to remove dicyclohexylurea byproducts and column chromatography using a DCM/methanol solvent system. Three different macro-CTA agents were prepared in total, MacroPEG
16-DMPA, MacroPEG
45-DMPA, and MacroPEG
114-DMPA, which varied in terms of the degree of polymerization of PEG chains. The chemical structure of the obtained Macro-PEG-DMPA was confirmed via proton nuclear magnetic resonance (
1H NMR) spectroscopy. The characteristic peaks corresponding to the ethylene oxide repeat units of PEG were observed in the region of 3.5 ppm, while the signals associated with the thiocarbonylthio moiety of DMPA were detected at approximately 1.2 and 3.3 ppm. These spectral features confirmed the successful attachment of the RAFT agent to the PEG backbone. In
Figure 1, the
1H NMR spectrum of MacroPEG
114-DMPA is shown.
RAFT Polymerization and Synthesis of PEGx-b-PVIMy Diblock Copolymers. Reversible Addition–Fragmentation Chain Transfer (RAFT) polymerization was employed to synthesize amphiphilic poly(ethylene glycol)-
block-poly(1-vinylimidazole) (PEG-
b-PVIM) copolymers, aiming to achieve precise control over molecular weight and narrow dispersity. However, initial polymerization attempts necessitated systematic optimization of reaction parameters, including the choice of chain transfer agent (CTA), solvent, monomer concentration, and initiator-to-CTA molar ratio, to attain successful polymerization. In the initial experiment, macroPEG
16 functionalized with 4-cyano-4-(dodecylsulfanylthiocarbonyl)sulfanyl pentanoic acid (macroPEG
16-CDPA) served as the CTA, with DMF as the solvent. The polymerization was conducted at 70 °C, utilizing a monomer concentration of 3.0 M and an AIBN-to-CTA molar ratio of 0.625. Under these conditions, no polymerization occurred, as confirmed by
1H NMR analysis. This lack of polymerization aligns with previous findings indicating that DMF may not effectively stabilize propagating radicals during RAFT polymerization of VIM [
12].
Subsequently, glacial acetic acid was selected as an alternative solvent due to its dual role in stabilizing propagating radicals and preventing undesired side reactions of the imidazole moieties [
17]. In the second experiment, macroPEG
16-CDPA was again employed as the CTA, with acetic acid as the solvent. The polymerization conditions included a monomer concentration of 2.0 M and an AIBN-to-CTA molar ratio of 1.0. Despite these adjustments, no polymerization was observed, suggesting that the selected CTA and initiator ratio were still unsuitable for effective polymerization. To investigate the effect of the CTA structure, the third experiment utilized CDPA without the macroPEG chain, while retaining acetic acid as the solvent. A monomer concentration of 2.5 M and an AIBN-to-CTA molar ratio of 0.5 were tested. Under these conditions, a monomer conversion of 61% was achieved, confirming that polymerization could proceed in the absence of the macroPEG segment. In the fourth experiment, the same conditions as the third were applied, except that macroPEG
16-CDPA was reintroduced as the CTA. Despite maintaining a monomer concentration of 2.5 M and an AIBN-to-CTA molar ratio of 0.5, a significant decrease in monomer conversion (15%) was observed. The lower monomer conversion in the presence of macroPEG
16-CDPA is likely due to steric hindrance and reduced reactivity caused by the bulky PEG
16 segment, which may limit the efficient transfer of the growing polymer chain between propagating radicals and the CTA.
In the fifth experiment, polymerization was performed using macroPEG16-CDPA in acetic acid with an increased monomer concentration of 4.0 M and an AIBN-to-CTA molar ratio of 0.625. Once again, no polymerization was detected, reinforcing the hypothesis that the selected CTA and reaction conditions were not optimal for efficient polymerization.
Given the limited success with the aforementioned CTAs, a new CTA was introduced in the subsequent phase of optimization. In the final experiment, PEG
16 functionalized with 2-dodecylthiocarbonylthio-2-methylpropanoic acid (macroPEG
16-DMPA) was employed as the CTA, maintaining a monomer concentration of 2.5 M and an AIBN-to-CTA molar ratio of 0.625. Under these conditions, a monomer conversion of 95% was achieved, indicating that macroPEG
16-DMPA is a more effective CTA for the RAFT polymerization of VIM.
Table 1 summarizes the experimental conditions and outcomes of the polymerization attempts.
After identifying the optimal conditions for VIM polymerization, the synthesis of five diblock copolymers with varying PEG-to-VIM molar ratios and different polymer chain lengths was carried out. The copolymers PEG
16-
b-PVIM
48, PEG
45-
b-PVIM
69, PEG
114-
b-VIM
86, PEG
45-
b-PVIM
34, and PEG
114-
b-VIM
48 were successfully synthesized under these optimized conditions. The successful preparation of these copolymers was confirmed via
1H NMR spectroscopy.
Figure 2 presents the
1H NMR spectrum of PEG
45-
b-PVIM
34 in D
2O. As observed in the spectrum, the characteristic aromatic peaks of the PVIM block appear in the region of 6.5–7.5 ppm, confirming the presence of the imidazole moieties within the polymer backbone. This further validates the successful polymerization and formation of the diblock copolymers.
(Co)polymer Molecular Weights and Compositions. In this study, five PEG-
b-PVIM diblock copolymers with varying chain lengths and compositions were synthesized under optimized RAFT polymerization conditions. The monomer conversion rates, determined via
1H NMR spectroscopy, and the molecular weights of the resulting copolymers are summarized in
Table 2.
As shown in
Table 2, monomer conversion rates ranged from 69.4% to 95.7%, with higher degrees of polymerization correlating with increased steric hindrance, which may have impeded monomer incorporation. Notably, the experimental M
n values obtained from GPC were consistently lower than the theoretical M
n values calculated based on monomer conversion determined by
1H NMR. This difference can be attributed to interactions between the copolymers and the GPC column, as well as potential micelle formation during analysis. Such interactions may lead to delayed elution, polymer aggregation, or altered hydrodynamic volume, ultimately resulting in underestimated molecular weights. Similar findings have been reported in the literature, where polymer–column interactions and self-assembly behaviors have caused GPC to underestimate molecular weights [
27,
28,
29].
The polydispersity indices (Ð) of the diblock copolymers, as determined by GPC, were all ≤1.3, indicating a narrow molecular weight distribution and a well-controlled polymerization process. These results suggest that factors such as polymer solubility, column interactions, and potential micellization may influence molecular weight determination by GPC.
Table 2 also presents the PVIM content of the diblock copolymers as calculated from
1H NMR spectroscopy. The experimental values closely matched the theoretical values, confirming that the polymerization of VIM was well-controlled. However, in all cases, the theoretical values calculated from initial monomer loading were higher than the experimental values, likely due to termination reactions occurring during polymerization.
Despite these variations, the successful synthesis of PEGx-b-PVIMy diblock copolymers with different PEG-to-PVIM ratios was confirmed via 1H NMR spectroscopy, further demonstrating the controlled nature of the polymerization process.
Effective pK Values. The effective pK values of the imidazole units in the synthesized diblock copolymers were determined using potentiometric titration. These values were calculated by analyzing the titration curves, where two asymptotic lines were constructed, and a perpendicular line was drawn to identify the equivalence point. The pH corresponding to half the NaOH volume (V) added at the equivalence point was recorded as the pK value.
As shown in
Table 3, the imidazole units in all copolymers exhibited pK values of approximately 4.2, irrespective of polymer composition. These values were lower than the reported pK of the free VIM monomer (6.07) [
30], which is consistent with the behavior of weak polyelectrolytes. This shift can be attributed to electrostatic repulsions between ionized imidazole groups along the polymer backbone, which hinder further ionization and result in a lower pK compared to the monomeric form.
Despite the deprotonation of the imidazole groups at physiological pH, the resulting copolymers remained water-soluble, as confirmed by acid–base titration.
Solution Micellization. To evaluate the self-assembly behavior of PEG
x-
b-PVIM
y diblock copolymers in aqueous solution, dynamic light scattering (DLS) measurements were performed to determine the hydrodynamic diameters (D
h) and polydispersity index (PDI) of the resulting aggregates. The thus-determined hydrodynamic diameters are listed in
Table 4. The same table shows the upper limit of the size of the micelles of the diblock copolymer calculated for fully stretched chains in spherical micelles, calculated by multiplying the total DP of the linear copolymers times 0.252 nm, the contribution of one monomer repeating unit [
31], then multiplying by two to convert the maximum micelle radius to the maximum diameter.
The effective hydrodynamic diameters were found to be 428.2 nm for PEG114-b-VIM86, 222 nm for PEG114-b-PVIM48, 243.2 nm for PEG45-b-PVIM69, and 370.4 nm for PEG45-b-PVIM34. The monomodal size distributions and PDI values below 0.2, and the higher values of experimental hydrodynamic diameters than the upper limit of the size of the micelles, suggest that relatively uniform aggregates were formed in all cases.
The formation of large aggregates instead of small micelles is consistent with previous studies on PEG-based block copolymers [
32]. Similar systems, such as PEG-
b-polyvinylpyrrolidone (PEG-
b-PVP) [
33], have been reported to self-assemble into nanoparticles or large aggregates in water, depending on the block ratio and polymer concentration. In the case of PEG
x-
b-VIM
y, the relatively high D
h values observed in our study suggest that these copolymers form aggregates through hydrophobic interactions between VIM blocks, which are not fully shielded by the hydrophilic PEG chains.
The monomodal size distributions observed in all samples indicate that the aggregates formed are relatively uniform, which differs from the behavior of some PEGylated diblock copolymers that exhibit bimodal or trimodal distributions depending on concentration and solvent conditions [
33]. This suggests that PEG
x-
b-PVIM
y copolymers may have a well-defined self-assembly pathway in aqueous media, where a single population of aggregates dominates under the studied conditions.
Overall, these findings indicate that PEG-b-VIM copolymers self-assemble into large aggregates in water rather than well-defined micelles, with the aggregate size influenced by the PEG-to-VIM ratio.
Conjugation of Diblock Copolymers PEG-b-PVIM with Chelating Agent NOTA. Following the successful synthesis of PEGx-b-PVIMy diblock copolymers, their conjugation with the maleimide-functionalized NOTA chelating agent was performed to facilitate radiolabeling for potential in vivo biodistribution studies. The conjugation reaction was carried out through a two-step aminolysis-click reaction mechanism. Initially, aminolysis of the trithiocarbonate end-group was performed using DBU, which acted as a nucleophile, attacking the electrophilic carbon of the trithiocarbonate moiety, leading to the formation of a free thiol group at the end of the polymer chain. This newly formed thiol then underwent a Michael addition (“click” reaction) with the electron-deficient maleimide functionality of NOTA, enabling efficient and selective bioconjugation under mild conditions. To ensure the removal of unreacted reagents and reaction byproducts, the conjugated PEG16-b-VIM48-NOTA copolymers were purified via precipitation in cold diethyl ether. The purified product was then collected by filtration, dried under vacuum, and stored for further characterization.
The successful conjugation of NOTA to PEG
16-
b-PVIM
48 was confirmed via
1H NMR spectroscopy, as shown in
Figure 3. The spectrum exhibited a distinct peak at 3.5 ppm, corresponding to the ethylene oxide protons of the PEG segment, while the aromatic protons of the imidazole moiety appeared around 7.0 ppm. Additionally, the aliphatic protons of NOTA were detected at approximately 2.8 ppm, providing clear evidence of successful modification.
This post-polymerization functionalization enhances the biomedical applicability of the synthesized diblock copolymers, enabling their use in radiolabeling and real-time tracking of nanocarriers in biological systems.
Radiolabeling. Radiolabeling was achieved using the 99mTc(CO)₃ tricarbonyl core. The precursor, fac-[99mTc(CO)3(H2O)3]+, was synthesized in-house by direct addition of 99mTcO4− to a sealed vial containing CO gas and NaBH4 as a reducing agent, followed by heating at 95 °C for 30 min. The reaction mixture was then adjusted to pH 7. Quality control by HPLC confirmed the efficient formation of the precursor, with a radiochemical purity exceeding 97%.
The macrocyclic compounds were radiolabeled with fac-[99mTc(CO)3(H2O)3]+ at low ligand concentrations, under heating at 80 °C for 30 min. HPLC analysis of the reaction mixtures revealed single peaks corresponding to each complex (99mTc-1 to 99mTc-7), eluting at 8–10 min, clearly separated from the precursor (3.5 min) and free pertechnetate (2.8 min). For clarity, the following labels are used to identify the radiolabeled compounds: Macro-PEG16 (99mTc-1), PEG16-b-PVIM48 (99mTc-2), PEG114-b-PVIM86 (99mTc-3), PEG114-b-PVIM48 (99mTc-4), PEG45-b-PVIM34 (99mTc-5), PEG45-b-PVIM69 (99mTc-6), and Macro-PEG45 (99mTc-7).
HPLC purification was performed to remove excess ligands and ensure high radiochemical purity before further in vitro and in vivo evaluation. Stability studies demonstrated that the radiotracers remained intact in their formulation for up to 6 h. Competitive incubation with histidine and cysteine—amino acids known to strongly coordinate the
fac-[
99mTc(CO)
3]
+ core—confirmed the high stability of the complexes, with >95% of the radiotracers remaining intact (
Table 5). In addition, incubation in human serum showed excellent stability over time. LogD
7.4 values indicated moderate lipophilicity (1.1–1.8).




 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}