
Synthesis and Properties of Photocurable Polymers Derived from the Polyesters of Glycerol and Aliphatic Dicarboxylic Acids

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
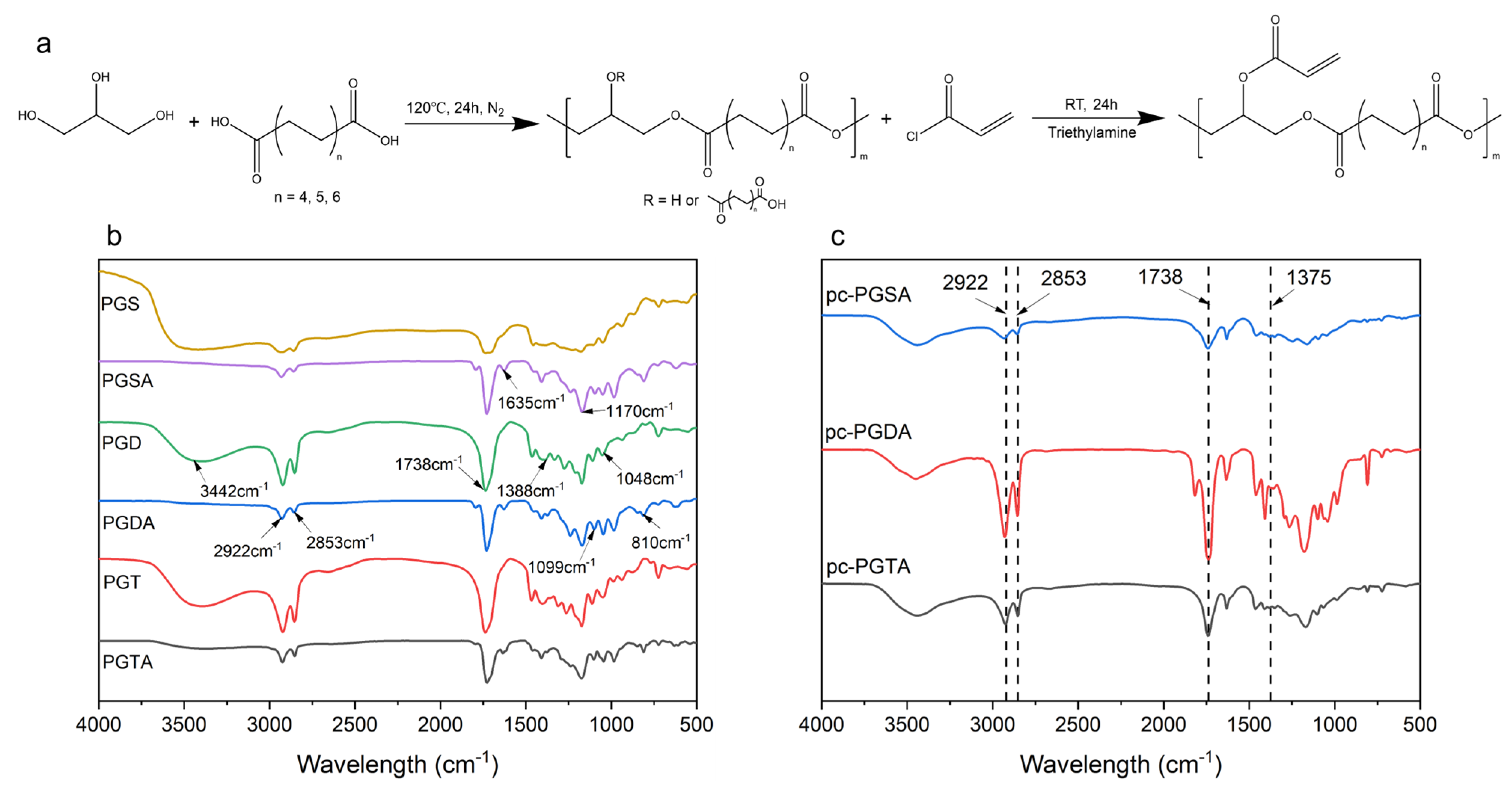
2.2. Synthesis of Polyglycerol Dicarboxylic Acid Esters
2.3. Acetylation of the Polyglycerol Dicarboxylic Acid Esters
2.4. Photocuring of the UV-Curable Polymers
2.5. Characterization of the Polymers and Photocured Elastomers
2.5.1. Fourier-Transform Infrared Spectroscopy (FT IR) Analysis
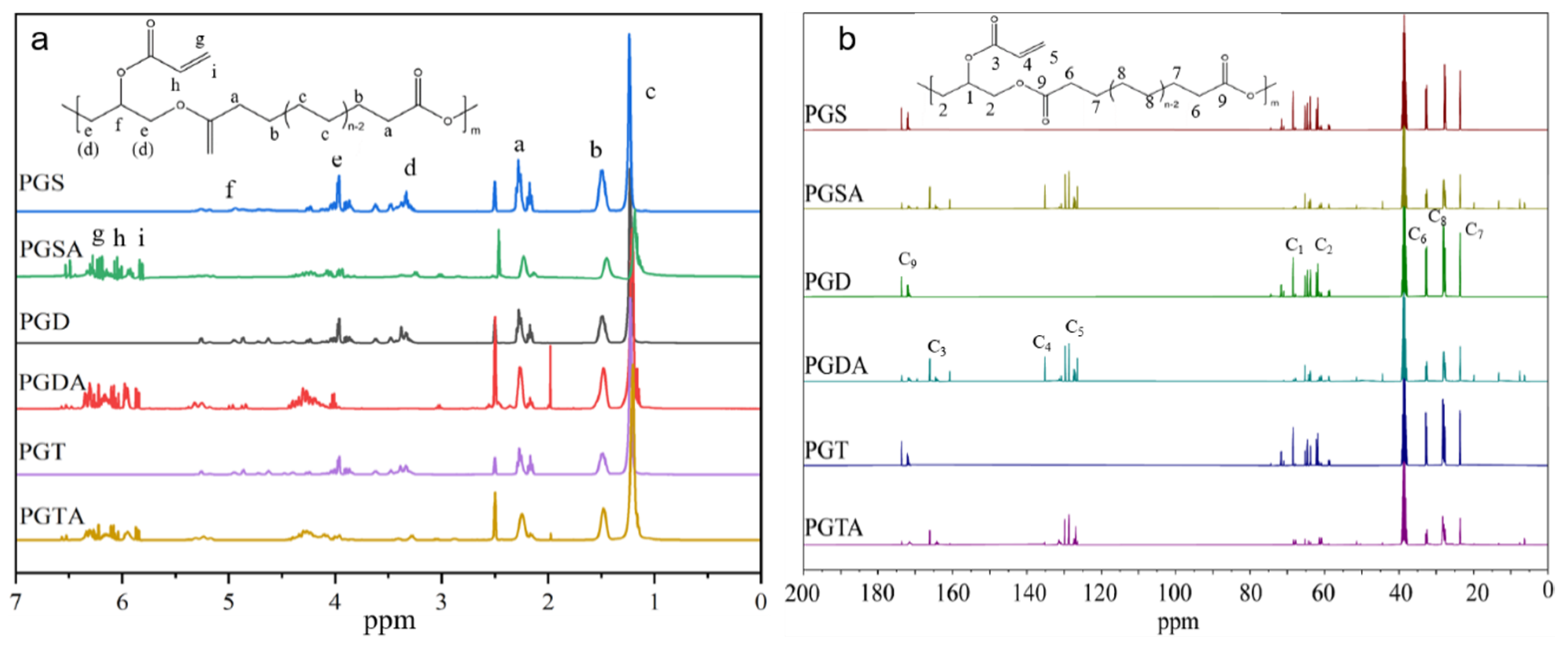
2.5.2. NMR Analysis
2.5.3. GPC Analysis
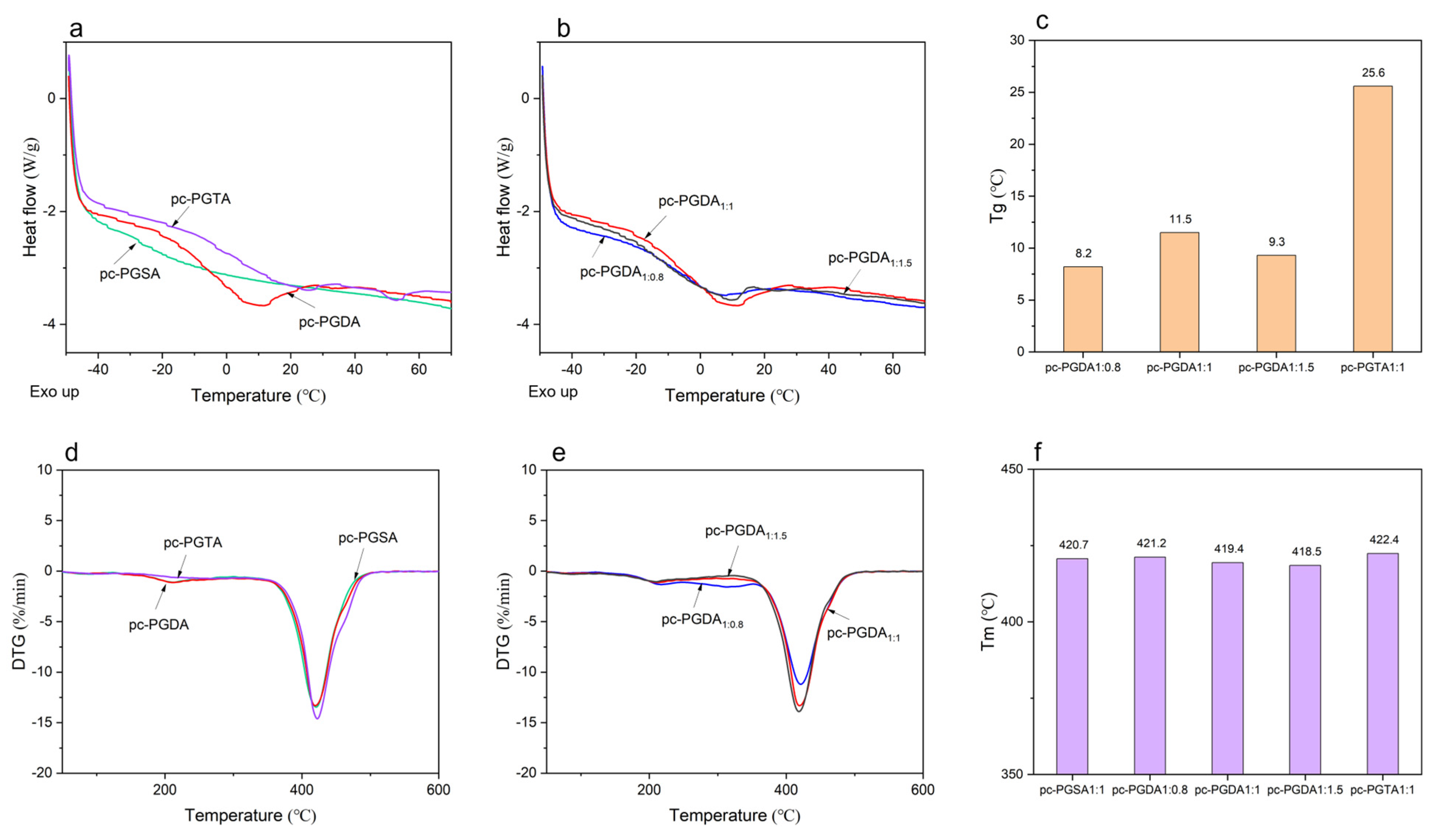
2.5.4. Differential Scanning Calorimetry (DSC) and Thermal Gravimetric (TGA) Analysis
2.5.5. Mechanical Property Analysis
2.5.6. Degradation Analysis
3. Results and Discussion
3.1. Effect of the Mole Ratio and Chain Lengths of ADCAs on the Prepolymers
3.2. Acetylation Efficiency of the Prepolymers
3.3. Photocuring Performance of the Acetylated Polyesters
3.4. The Properties of Photocured Elastomers
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Randhawa, A.; Dutta, S.D.; Ganguly, K.; Patel, D.K.; Patil, T.V.; Lim, K.-T. Recent advances in 3D printing of photocurable polymers: Types, mechanism, and tissue engineering application. Macromol. Biosci. 2023, 23, 2200278. [Google Scholar] [CrossRef]
- Crivello, J.V. UV and electron beam-induced cationic polymerization. Nucl. Instrum. Methods Phys. Res. 1999, 151, 8–21. [Google Scholar] [CrossRef]
- Kim, J.-S.; Noh, S.-T.; Kweon, J.-O.; Cho, B.-S. Photopolymerization kinetic studies of UV-curable sulfur-containing difunctional acrylate monomers using photo-DSC. Macromol. Res. 2015, 23, 341–349. [Google Scholar] [CrossRef]
- Bretterbauer, K.; Holzmann, C.; Rubatscher, E.; Schwarzinger, C.; Roessler, A.; Paulik, C. UV-curable coatings of highly crosslinked trimethylmelamine based acrylates and methacrylates. Eur. Polym. J. 2013, 49, 4141–4148. [Google Scholar] [CrossRef]
- Zheng, C.; Liu, G.; Hu, H. UV-curable antismudge coatings. ACS Appl. Mater. Interfaces 2017, 9, 25623–25630. [Google Scholar] [CrossRef]
- Hakeim, O.A.; Arafa, A.A.; Zahran, M.K.; Abdou, L.A.W. Characterisation and application of pigmented UV-curable inkjet inks. Pigment Resin Technol. 2018, 47, 164–172. [Google Scholar] [CrossRef]
- Sugita, H.; Itou, K.; Itou, Y.; Wada, N.; Kurita, T.U.S.; Hirose, Y.; Hatase, K.; Matsumoto, H.; Ichinohe, D. Multi-acrylate-based UV-curable dismantlable adhesives. Int. J. Adhes. Adhes. 2021, 104, 102758. [Google Scholar] [CrossRef]
- Burke, G.; Devine, D.M.; Major, I. Effect of stereolithography 3D printing on the properties of PEGDMA hydrogels. Polymers 2020, 12, 2015. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, F.; Shi, Q.; Zhang, J.; Xiang, Z.; Li, N.; Huang, X.; Song, J. Three-dimensional printing chitosan-based bolus used for radiotherapy. ACS Appl. Bio Mater. 2021, 4, 7094–7102. [Google Scholar] [CrossRef]
- Rajput, M.; Mondal, P.; Yadav, P.; Chatterjee, K. Light-based 3D bioprinting of bone tissue scaffolds with tunable mechanical properties and architecture from photocurable silk fibroin. Int. J. Biol. Macromol. 2022, 202, 644–656. [Google Scholar] [CrossRef]
- Wang, M.; Yang, F.; Luo, H.; Jiang, Y.; Zhuang, K.; Tan, L. Photocuring and gelatin-based antibacterial hydrogel for skin care. Biomacromolecules 2023, 24, 4218–4228. [Google Scholar] [CrossRef]
- Fu, K.; Wang, Y.; Yan, C.; Yao, Y.; Chen, Y.; Dai, J.; Lacey, S.; Wang, Y.; Wan, J.; Li, T.; et al. Graphene oxide-based electrode inks for 3D-printed lithium-ion batteries. Adv. Mater. 2016, 28, 2587–2594. [Google Scholar] [CrossRef]
- Wu, P.; Wang, J.; Wang, X. A critical review of the use of 3-D printing in the construction industry. Autom. Constr. 2016, 68, 21–31. [Google Scholar] [CrossRef]
- Quan, H.; Zhang, T.; Xu, H.; Luo, S.; Nie, J.; Zhu, X. Photo-curing 3D printing technique and its challenges. Bioact. Mater. 2020, 5, 110–115. [Google Scholar] [CrossRef]
- Kim, W.G.; Lee, J.Y. Cure properties of methacrylate-type prepolymer that include cyclohexane moiety. J. Appl. Polym. Sci. 2004, 92, 43–52. [Google Scholar] [CrossRef]
- Zhang, B.; Li, S.; Hingorani, H.; Serjouei, A.; Larush, L.; Pawar, A.A.; Goh, W.H.; Sakhaei, A.H.; Hashimoto, M.; Kowsari, K.; et al. Highly stretchable hydrogels for UV curing based high-resolution multimaterial 3D printing. J. Mater. Chem. B 2018, 6, 3246–3253. [Google Scholar] [CrossRef]
- Zhao, T.; Yu, R.; Li, S.; Li, X.; Zhang, Y.; Yang, X.; Zhao, X.; Wang, C.; Liu, Z.; Dou, R.; et al. Superstretchable and processable silicone elastomers by digital light processing 3D printing. ACS Appl. Mater. Interfaces 2019, 11, 14391–14398. [Google Scholar] [CrossRef] [PubMed]
- Iedema, P.D.; Schamböck, V.; Boonen, H.; van der Linden, M.N.; Willemse, R. Photocuring of di-Acrylate in presence of oxygen. Chem. Eng. Sci. 2019, 207, 130–144. [Google Scholar] [CrossRef]
- Ge, Q.; Sakhaei, A.H.; Lee, H.; Dunn, C.K.; Fang, N.X.; Dunn, M.L. Multimaterial 4D printing with tailorable shape memory polymers. Sci. Rep. 2016, 6, 31110. [Google Scholar] [CrossRef]
- Arcaute, K.; Mann, B.; Wicker, R. Stereolithography of spatially controlled multi-material bioactive poly(ethylene glycol) scaffolds. Acta Biomater. 2010, 6, 1047–1054. [Google Scholar] [CrossRef]
- Ji, Z.; Zhang, X.; Yan, C.; Jia, X.; Xia, Y.; Wang, X.; Zhou, F. 3D printing of photocuring elastomers with excellent mechanical strength and resilience. Macromol. Rapid Commun. 2019, 40, 1800873. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.K.; Sakhaei, A.H.; Layani, M.; Zhang, B.; Ge, Q.; Magdassi, S. Highly stretchable and UV curable elastomers for digital light processing based 3D printing. Adv. Mater. 2017, 29, 1606000. [Google Scholar] [CrossRef] [PubMed]
- Marques, D.S.; Santos, J.M.C.; Ferreira, P.; Correia, T.R.; Correia, I.J.; Gil, M.H.; Baptista, C.M.S.G. Functionalization and photocuring of an L-lactic acid macromer for biomedical applications. Int. J. Polym. Mater. Polym. Biomater. 2016, 65, 497–507. [Google Scholar] [CrossRef]
- Wang, S.; Dai, J.; Teng, N.; Hu, J.; Zhao, W.; Liu, X. Synthesis of mechanically robust and self-healing UV-curable materials from renewable feedstock. ACS Sustain. Chem. Eng. 2020, 8, 16842–16852. [Google Scholar] [CrossRef]
- Hegde, M.; Meenakshisundaram, V.; Chartrain, N.; Sekhar, S.; Tafti, D.; Williams, C.B.; Long, T.E. 3D Printing all-aromatic polyimides using mask-projection stereolithography: Processing the nonprocessable. Adv. Mater. 2017, 29, 1701240. [Google Scholar] [CrossRef] [PubMed]
- Field, D.E.; Griffith, J.R. Cross-linked fluoropolymer coatings. Ind. Eng. Chem. Prod. Res. Dev. 1975, 14, 52–54. [Google Scholar] [CrossRef]
- Ge, Q.; Chen, Z.; Cheng, J.; Zhang, B.; Zhang, Y.-F.; Li, H.; He, X.; Yuan, C.; Liu, J.; Magdassi, S.; et al. 3D printing of highly stretchable hydrogel with diverse UV curable polymers. Sci. Adv. 2021, 7, 4261. [Google Scholar] [CrossRef] [PubMed]
- Si, Z.; Li, J.; Ma, L.; Cai, D.; Li, S.; Baeyens, J.; Degrève, J.; Nie, J.; Tan, T.; Qin, P. The ultrafast and continuous fabrication of a polydimethylsiloxane membrane by ultraviolet-induced polymerization. Angew. Chem. Int. Ed. 2019, 131, 17175–17179. [Google Scholar] [CrossRef] [PubMed]
- Şabani, S.; Önen, A.H.; Güngör, A. Preparation of hyperbranched polyester polyol-based urethane acrylates and applications on UV-curable wood coatings. J. Coat. Technol. Res. 2012, 9, 703–716. [Google Scholar] [CrossRef]
- Liu, Y.; Lin, Y.; Jiao, T.; Lu, G.; Liu, J. Photocurable modification of inorganic fillers and their application in photopolymers for 3D printing. Polym. Chem. 2019, 10, 6324–6333. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, J.; An, Z.; Kankala, R.K.; Chen, A.-Z.; Wang, S.-B.; Li, Y. Photocuring 3D printable self-healing polymers. Eur. Polym. J. 2023, 199, 112471. [Google Scholar] [CrossRef]
- Wu, H.-J.; Hu, M.-H.; Tuan-Mu, H.-Y.; Hu, J.-J. Preparation of aligned poly(glycerol sebacate) fibrous membranes for anisotropic tissue engineering. Mater. Sci. Eng. C 2019, 100, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Heydari, P.; Parham, S.; Kharazi, A.Z.; Javanmard, S.H.; Asgary, S. In vitro comparison study of plasma treated bilayer PGS/PCL and PGS/PLA scaffolds for vascular tissue engineering. Fibers Polym. 2022, 23, 2384–2393. [Google Scholar] [CrossRef]
- Migneco, F.; Huang, Y.C.; Birla, R.K.; Hollister, S.J. Poly(glycerol-dodecanoate), a biodegradable polyester for medical devices and tissue engineering scaffolds. Biomaterials 2009, 30, 6479–6484. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Huang, Y.-C. Electrospun fibrous scaffolds of Poly (glycerol-dodecanedioate) for engineering neural tissues from mouse embryonic stem cells. J. Vis. Exp. 2014, 88, e51587. [Google Scholar]
- Ramaraju, H.; Solorio, L.D.; Bocks, M.L.; Hollister, S.J. Degradation properties of a biodegradable shape memory elastomer, poly(glycerol dodecanoate), for soft tissue repair. PLoS ONE 2020, 15, e0229112. [Google Scholar] [CrossRef] [PubMed]
- Ramaraju, H.; Massarella, D.; Wong, C.; Verga, A.S.; Kish, E.C.; Bocks, M.L.; Hollister, S.J. Percutaneous delivery and degradation of a shape memory elastomer poly(glycerol dodecanedioate) in porcine pulmonary arteries. Biomaterials 2023, 293, 121950. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Deng, H.; SKenderes, M.; Su, J.-W.; Whittington, A.G.; Lin, J. Chemically interconnected thermotropic polymers for transparency-tunable and impact-resistant windows. ACS Appl. Mater. Interfaces 2019, 11, 5393–5400. [Google Scholar] [CrossRef]
- Nijst, C.L.E.; Bruggeman, J.P.; Karp, J.M.; Ferreira, L.; Zumbuehl, A.; Bettinger, C.J.; Langer, R. Synthesis and characterization of photocurable elastomers from poly(glycerol-co-sebacate). Biomacromolecules 2007, 8, 3067–3073. [Google Scholar] [CrossRef]
- Yeh, Y.-C.; Highley, C.B.; Ouyang, L.; Burdick, J.A. 3D printing of photocurable poly(glycerol sebacate) elastomers. Biofabrication 2016, 8, 045004. [Google Scholar] [CrossRef]
- Akman, R.; Ramaraju, H.; Hollister, S.J. Development of photocrosslinked poly(glycerol dodecanedioate), a biodegradable shape memory polymer for 3D printed tissue engineering applications. Adv. Eng. Mater. 2021, 23, 2100219. [Google Scholar] [CrossRef]
- Ramaraju, H.; Ul-Haque, A.; Verga, A.S.; Bocks, M.L.; Hollister, S.J. Modulating nonlinear elastic behavior of biodegradable shape memory elastomer and small intestinal submucosa (SIS) composites for soft tissue repair. J. Mech. Behav. Biomed. Mater. 2020, 110, 103965. [Google Scholar] [CrossRef] [PubMed]
- Martín-Cabezuelo, R.; Vilariño-Feltrer, G.; Vallés-Lluch, A. Influence of pre-polymerisation atmosphere on the properties of pre- and poly(glycerol sebacate). Mater. Sci. Eng. C 2021, 119, 111429. [Google Scholar] [CrossRef] [PubMed]
- Celli, A.; Marchese, P.; Sullalti, S.; Berti, C.; Barbiroli, G.; Commereuc, S.; Verney, V. Preparation of new biobased polyesters containing glycerol and their photodurability for outdoor applications. Green Chem. 2012, 14, 182–187. [Google Scholar] [CrossRef]
- Perin, G.B.; Felisberti, M.I. Polyesters inspired by glycerides: Enzymatic polycondensation, structure, properties, and nanoparticle preparation. Macromolecules 2023, 56, 6968–6977. [Google Scholar] [CrossRef]
- Yin, B.; Zhang, J. A novel photocurable modified epoxy resin for high heat resistance coatings. Colloid Polym. Sci. 2020, 298, 1303–1312. [Google Scholar] [CrossRef]
- Wei, D.; Liao, B.; Yong, Q.; Li, T.; Wang, H.; Huang, J.; Pang, H. Castor oil based hyperbranched urethane acrylates and their performance as UV-curable coatings. J. Macromol. Sci. Part A 2018, 55, 422–432. [Google Scholar] [CrossRef]
- Xu, H.; Qiu, F.; Wang, Y.; Wu, W.; Yang, D.; Guo, Q. UV-curable waterborne polyurethane-acrylate: Preparation, characterization and properties. Prog. Org. Coat. 2012, 73, 47–53. [Google Scholar] [CrossRef]
- Voet, V.S.D.; Strating, T.; Schnelting, G.H.M.; Dijkstra, P.; Tietema, M.; Xu, J.; Woortman, A.J.J.; Loos, K.; Jager, J.; Folkersma, R. Biobased acrylate photocurable resin formulation for stereolithography 3D printing. ACS Omega 2018, 3, 1403–1408. [Google Scholar] [CrossRef]
- Akman, R.; Ramaraju, H.; Verga, A.; Hollister, S.J. Multimodal 3D printing of biodegradable shape memory elastomer resins for patient specific soft tissue repair. Appl. Mater. Today 2022, 29, 101666. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prepolymers | PGS | PGD | PGT | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ADCA | Sebacic acid | Dodecanedioic acid | Tetradecanedioic acid | |||||||
| Gly:ADCA | 1:1 | 1:0.8 | 1:1 | 1:1.5 | 1:1 | |||||
| Reaction time (h) | 24 | 48 | 24 | 48 | 24 | 48 | 24 | 48 | 24 | 48 |
| Mn (g/mol) | 325 | 410 | 291 | 394 | 346 | 475 | 328 | 505 | 407 | 599 |
| Mw(g/mol) | 1707 | 2356 | 1782 | 1813 | 1865 | 2712 | 1937 | 2954 | 2188 | 5695 |
| PDI | 5.25 | 5.75 | 6.12 | 4.60 | 5.39 | 5.71 | 5.91 | 5.85 | 5.38 | 9.51 |
| p-OH/s-OH | 0.92 | 0.85 | 1.05 | 1.02 | 0.85 | 0.77 | 0.73 | 0.69 | 0.86 | 0.70 |
| ED1 | 0.43 | 0.45 | 0.32 | 0.37 | 0.39 | 0.44 | 0.43 | 0.47 | 0.39 | 0.43 |
| ED2 | 0.23 | 0.27 | 0.16 | 0.19 | 0.23 | 0.26 | 0.28 | 0.29 | 0.22 | 0.26 |
| –C=O a | 1.83 | 1.41 | 1.80 | 2.55 | 1.77 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, R.; Yao, W.; Fu, Y.; Lu, F.; Chen, X. Synthesis and Properties of Photocurable Polymers Derived from the Polyesters of Glycerol and Aliphatic Dicarboxylic Acids. Polymers 2024, 16, 1278. https://doi.org/10.3390/polym16091278
Hu R, Yao W, Fu Y, Lu F, Chen X. Synthesis and Properties of Photocurable Polymers Derived from the Polyesters of Glycerol and Aliphatic Dicarboxylic Acids. Polymers. 2024; 16(9):1278. https://doi.org/10.3390/polym16091278
Chicago/Turabian StyleHu, Rui, Weipeng Yao, Yingjuan Fu, Fuyuan Lu, and Xiaoqian Chen. 2024. "Synthesis and Properties of Photocurable Polymers Derived from the Polyesters of Glycerol and Aliphatic Dicarboxylic Acids" Polymers 16, no. 9: 1278. https://doi.org/10.3390/polym16091278
APA StyleHu, R., Yao, W., Fu, Y., Lu, F., & Chen, X. (2024). Synthesis and Properties of Photocurable Polymers Derived from the Polyesters of Glycerol and Aliphatic Dicarboxylic Acids. Polymers, 16(9), 1278. https://doi.org/10.3390/polym16091278