

Efficient Alcoholysis of Poly(ethylene terephthalate) by Using Supercritical Carbon Dioxide as a Green Solvent


 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Process
2.3. Characterization
2.4. Simulation Details
2.5. Self-Diffusion Coefficient
2.6. Hydrogen Bonds
3. Result and Discussion
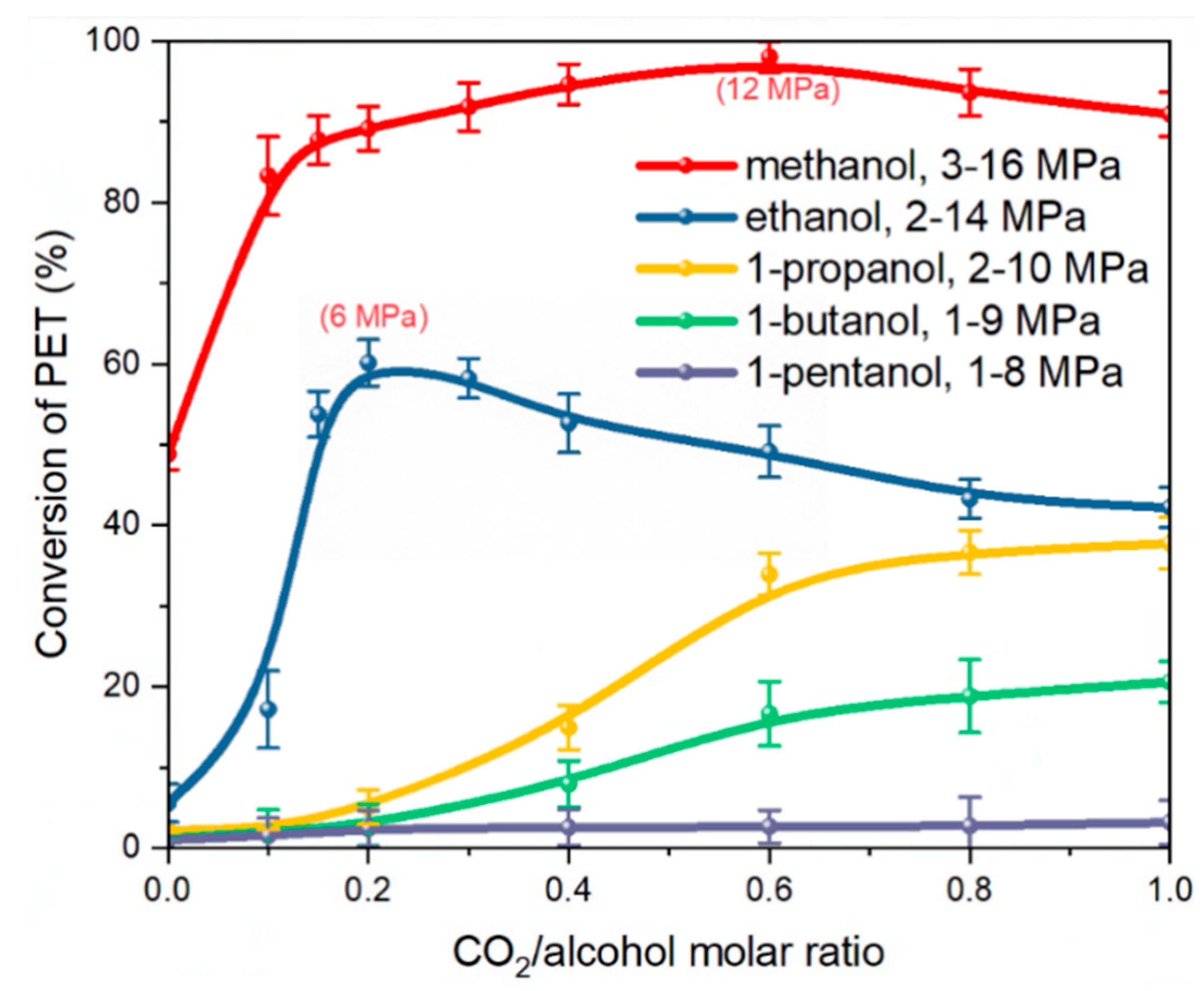
3.1. Effect of CO2-Alcohols Molar Ratio on PET Depolymerization Behavior
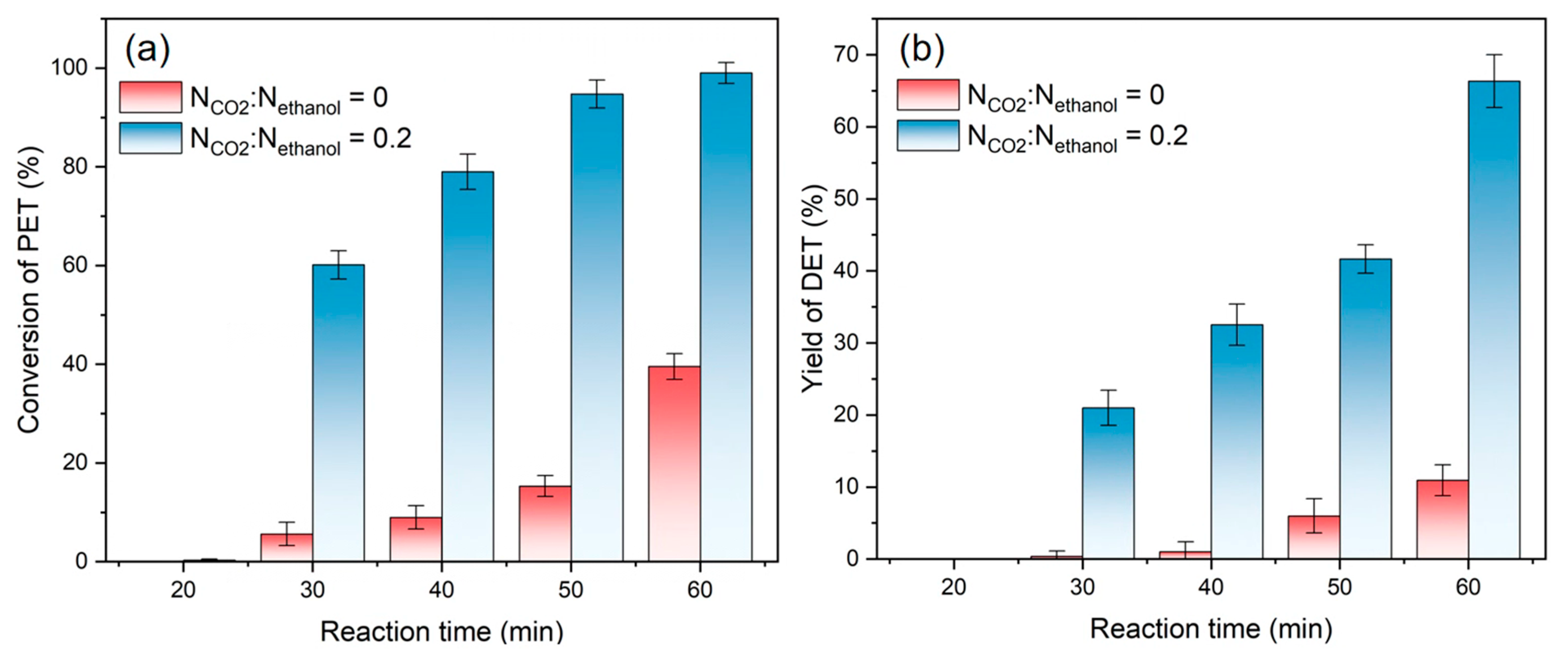
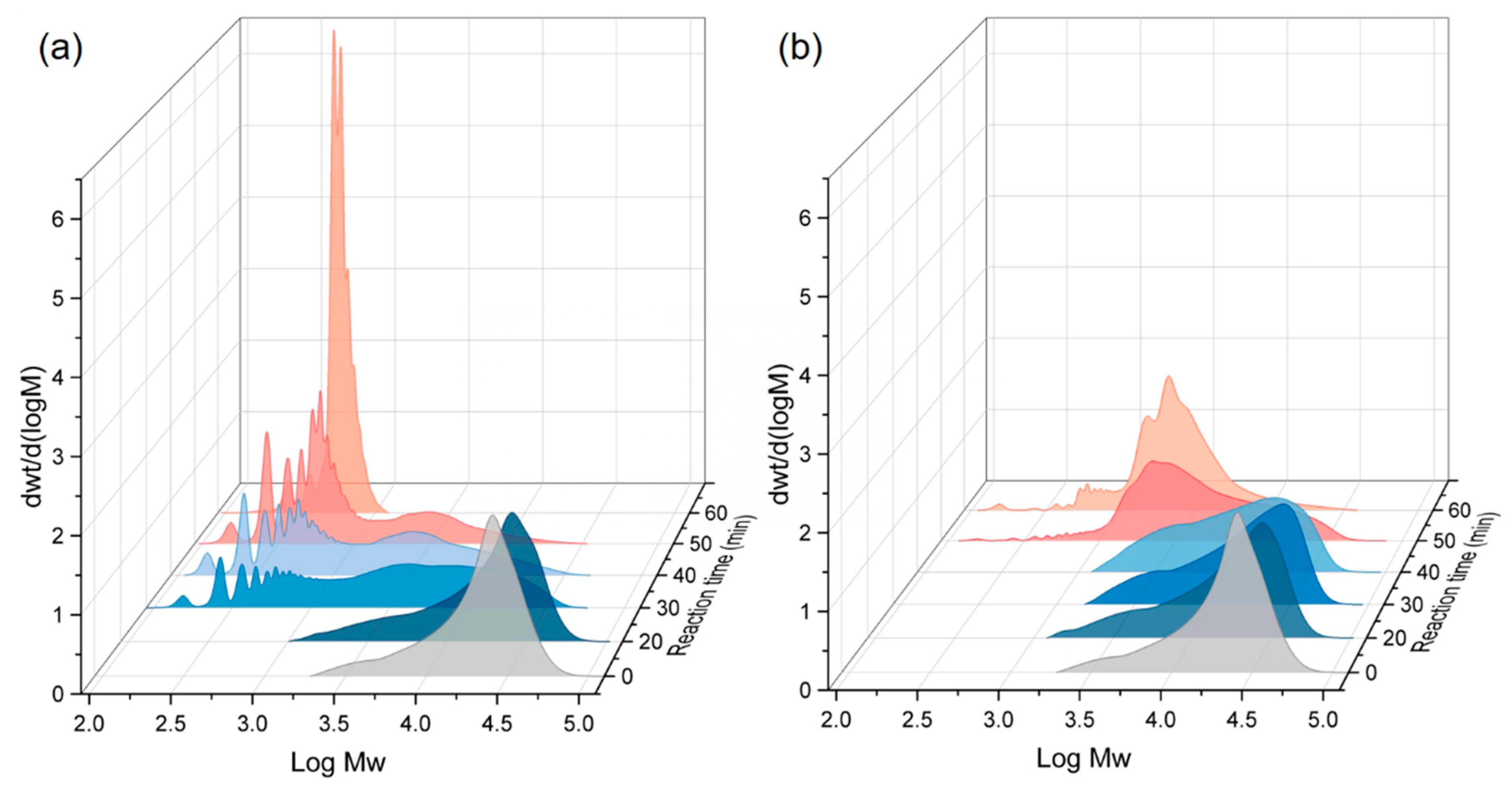
3.2. Effect of the Reaction Time on Depolymerization with CO2 and Ethanol
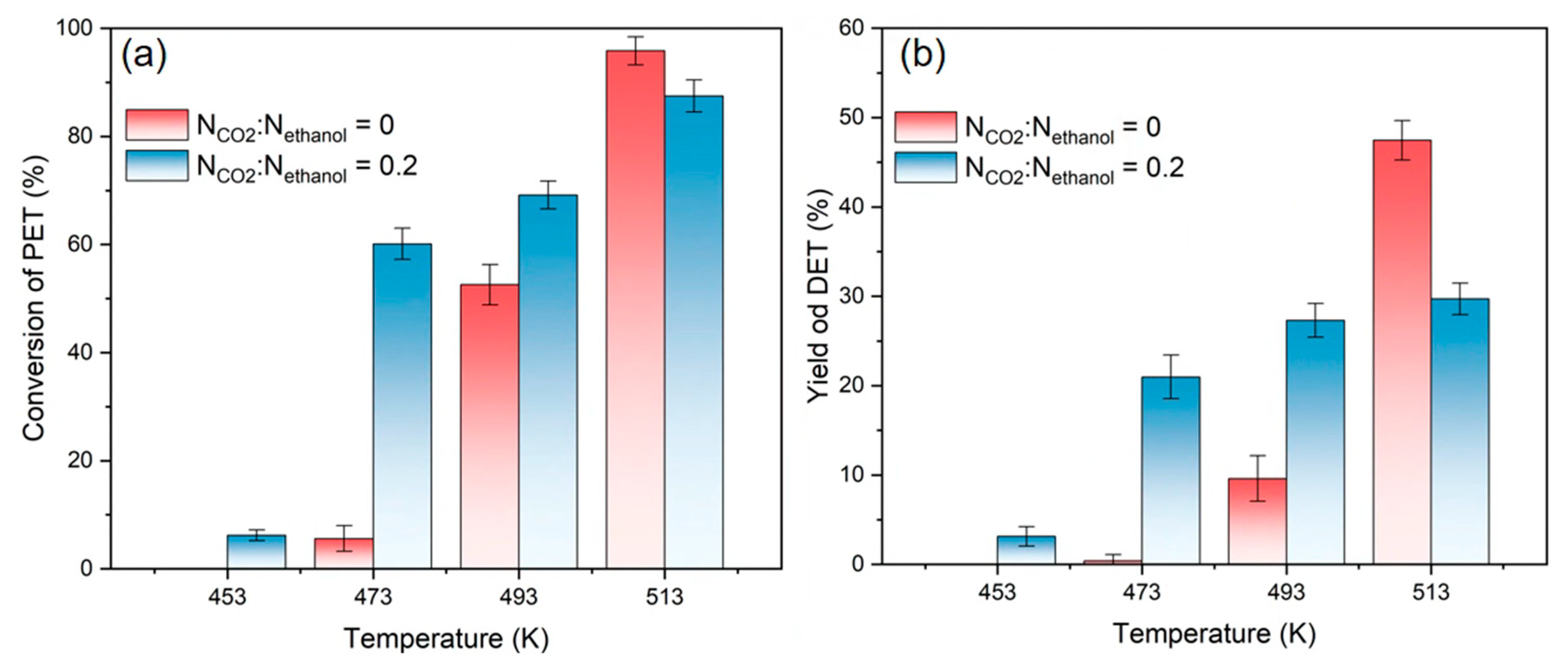
3.3. Effect of the Reaction Temperature on Depolymerization
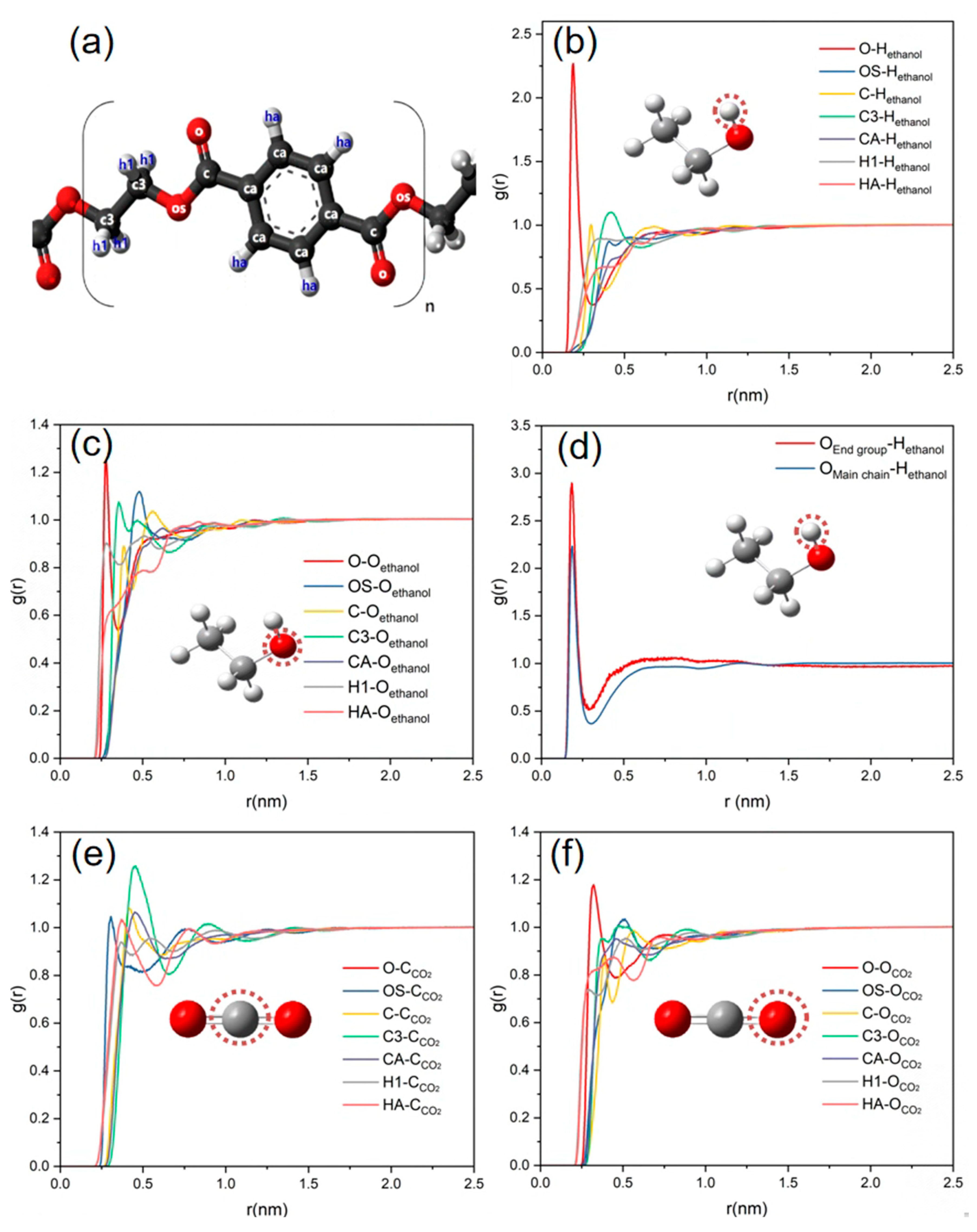
3.4. Intermolecular Interactions in Ternary Systems of PET-Alcohol-CO2
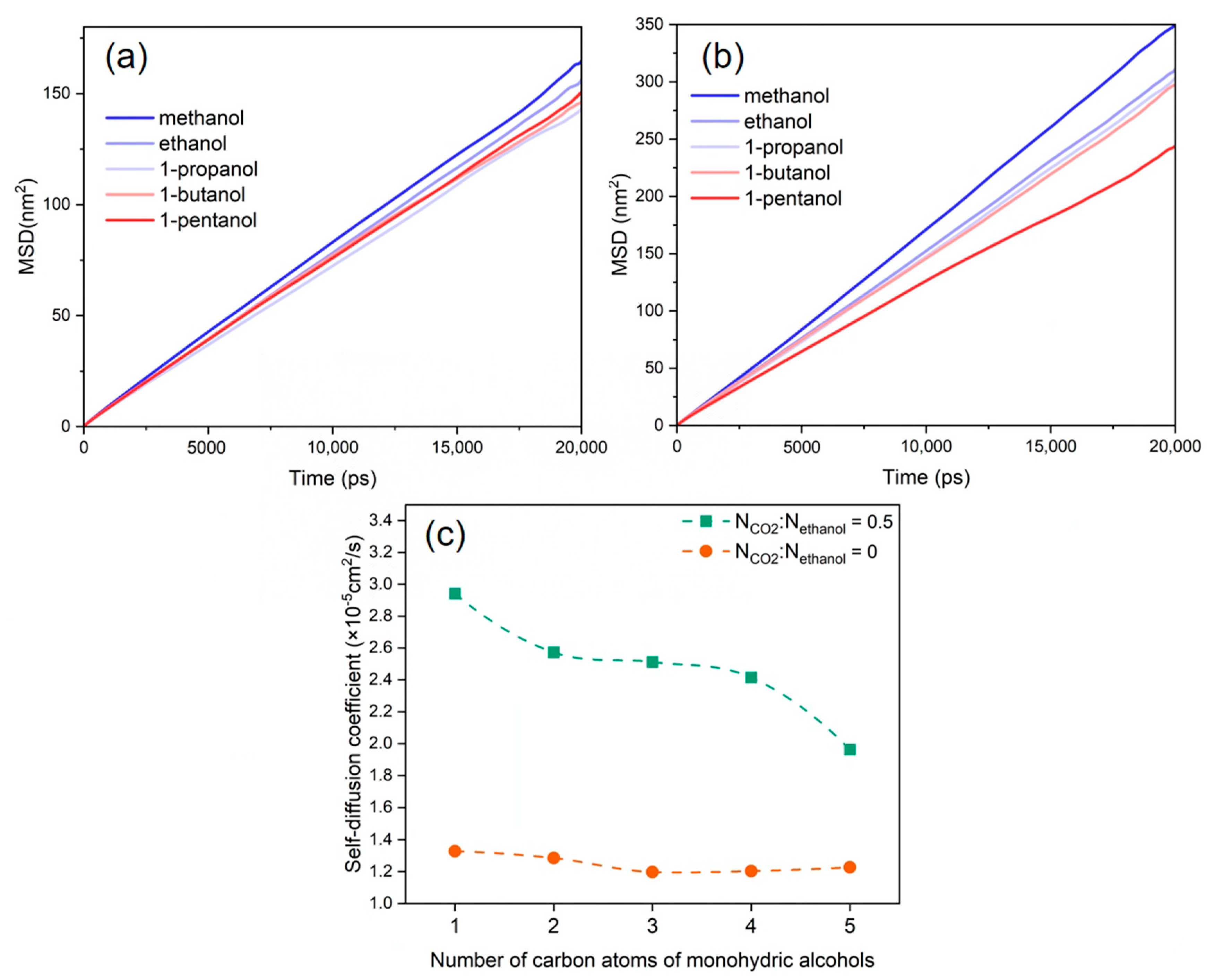
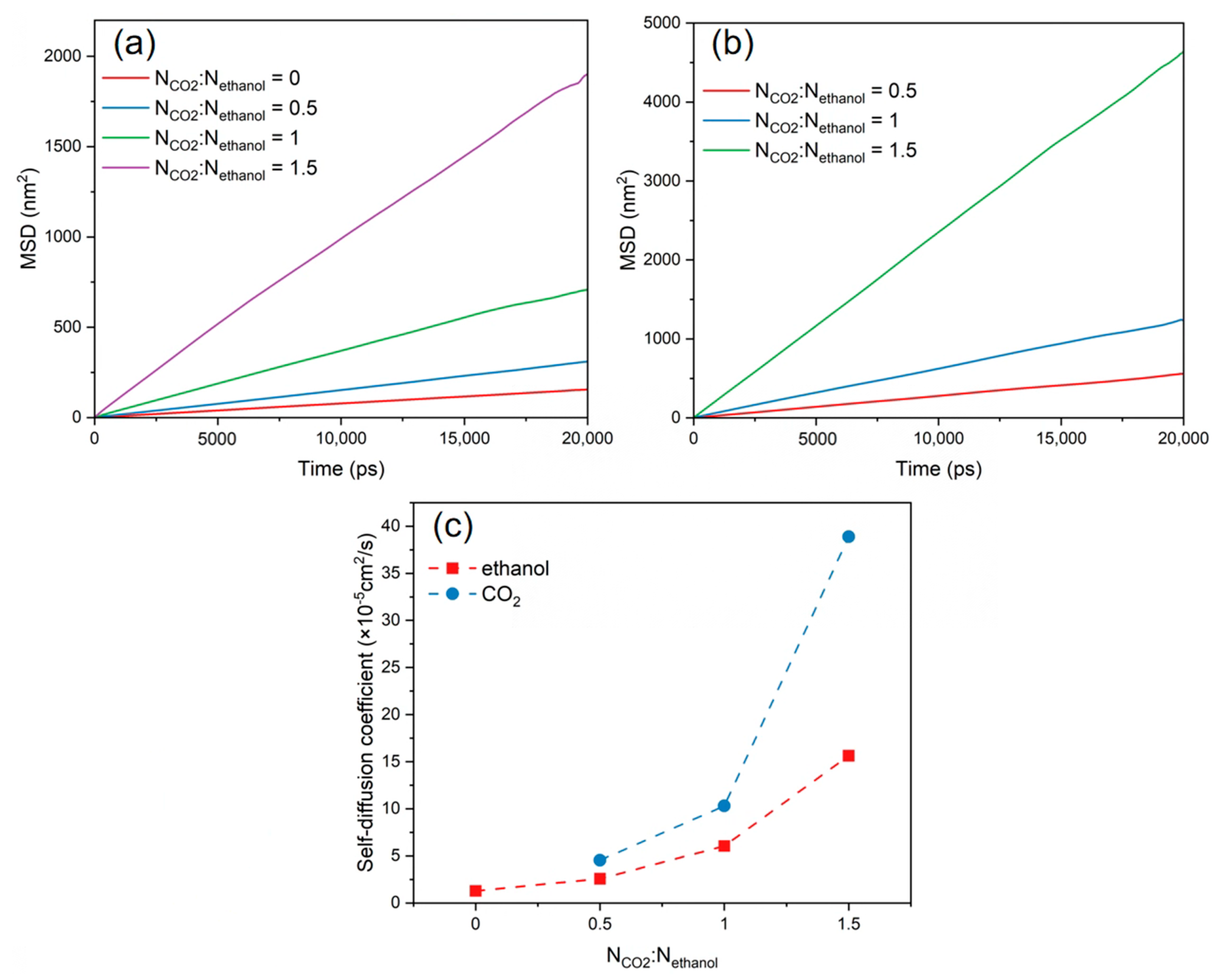
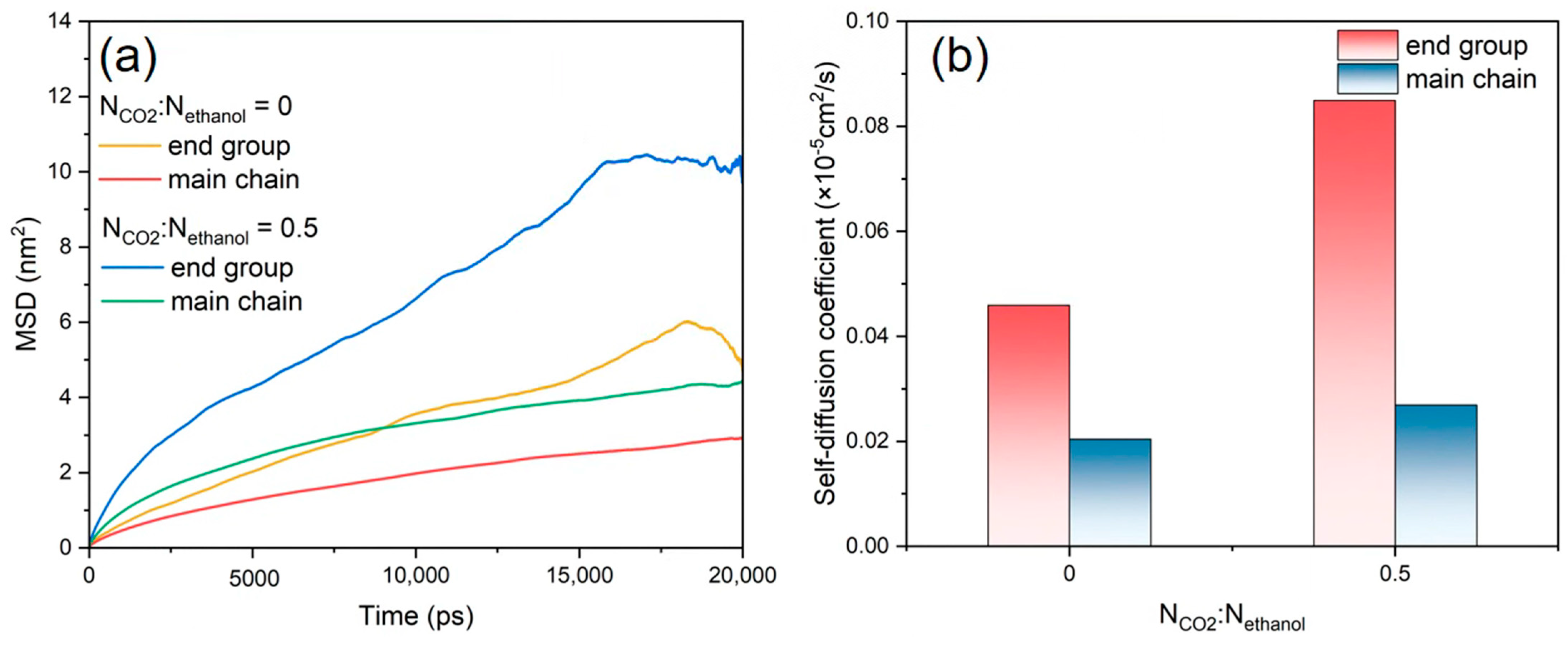
3.5. Diffusion Behaviour
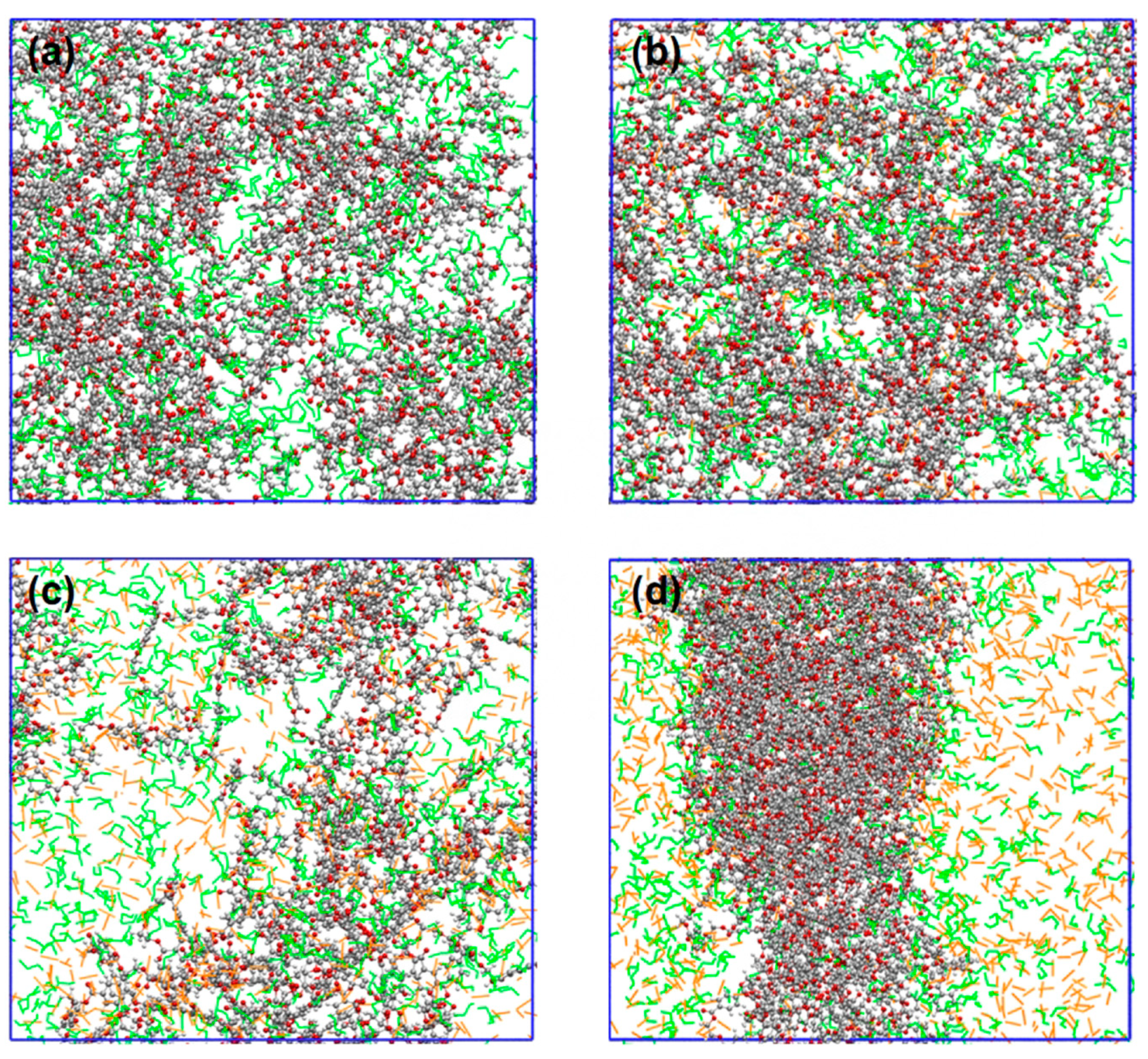
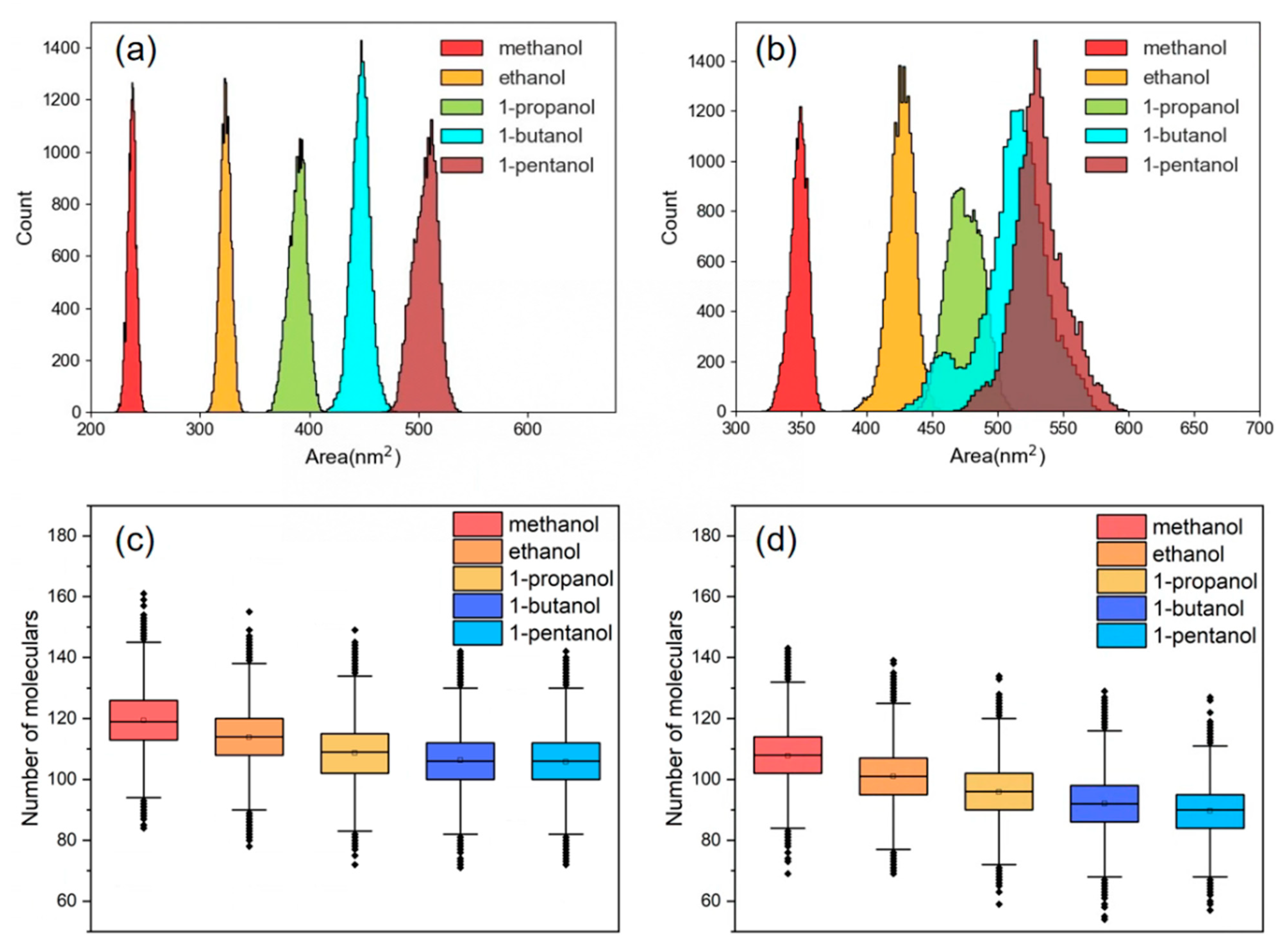
3.6. Swelling Behaviors
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, Y.; Liu, S.; Wang, P.; Jian, X.; Liao, X.; Chen, W.-Q. China’s Roadmap to Plastic Waste Management and Associated Economic Costs. J. Environ. Manag. 2022, 309, 114686. [Google Scholar] [CrossRef] [PubMed]
- Nisticò, R. Polyethylene Terephthalate (PET) in the Packaging Industry. Polym. Test. 2020, 90, 106707. [Google Scholar] [CrossRef]
- IHS Markit. Plasticizers. Chemical Economics Handbook; IHS Markit Ltd.: London, UK, 2018; Available online: https://www.spglobal.com/commodityinsights/en/ci/products/plasticizers-chemical-economics-handbook.html (accessed on 1 April 2024).
- Korley, L.T.J.; Epps, T.H.; Helms, B.A.; Ryan, A.J. Toward Polymer Upcycling—Adding Value and Tackling Circularity. Science 2021, 373, 66–69. [Google Scholar] [CrossRef]
- Nandi, S.; Mahish, S.S.; Das, S.K.; Datta, M.; Nath, D. A Review of Various Recycling Methods of PET Waste: An Avenue to Circularity. Polym.-Plast. Technol. Mater. 2023, 62, 1663–1683. [Google Scholar] [CrossRef]
- Chen, R.; Deng, S.; Cui, T.; Duan, S.; Jia, Q.; Zhang, L. Progress in Recycling and Reutilization of Waste Polyethylene Terephthalate. Prog. Rubber Plast. Recycl. Technol. 2024, 40, 77–97. [Google Scholar] [CrossRef]
- Martín, A.J.; Mondelli, C.; Jaydev, S.D.; Pérez-Ramírez, J. Catalytic Processing of Plastic Waste on the Rise. Chem 2021, 7, 1487–1533. [Google Scholar] [CrossRef]
- Jehanno, C.; Alty, J.W.; Roosen, M.; De Meester, S.; Dove, A.P.; Chen, E.Y.-X.; Leibfarth, F.A.; Sardon, H. Critical Advances and Future Opportunities in Upcycling Commodity Polymers. Nature 2022, 603, 803–814. [Google Scholar] [CrossRef]
- Barnard, E.; Rubio Arias, J.J.; Thielemans, W. Chemolytic Depolymerisation of PET: A Review. Green Chem. 2021, 23, 3765–3789. [Google Scholar] [CrossRef]
- Goto, M. Chemical Recycling of Plastics Using Sub- and Supercritical Fluids. J. Supercrit. Fluids 2009, 47, 500–507. [Google Scholar] [CrossRef]
- Goto, M.; Jin, F.; Zhou, Q.; Wu, B. Supercritical Water Process for the Chemical Recycling of Waste Plastics. AIP Conf. Proc. 2010, 1251, 169–172. [Google Scholar] [CrossRef]
- Sako, T.; Sugeta, T.; Otake, K.; Nakazawa, N.; Sato, M.; Namiki, K.; Tsugumi, M. Depolymerization of Polyethylene Terephthalate to Monomers with Supercritical Methanol. J. Chem. Eng. Jpn. JCEJ 1997, 30, 342–346. [Google Scholar] [CrossRef]
- De Castro, R.E.N.; Vidotti, G.J.; Rubira, A.F.; Muniz, E.C. Depolymerization of Poly(Ethylene Terephthalate) Wastes Using Ethanol and Ethanol/Water in Supercritical Conditions. J. Appl. Polym. Sci. 2006, 101, 2009–2016. [Google Scholar] [CrossRef]
- Lozano-Martinez, P.; Torres-Zapata, T.; Martin-Sanchez, N. Directing Depolymerization of PET with Subcritical and Supercritical Ethanol to Different Monomers through Changes in Operation Conditions. ACS Sustain. Chem. Eng. 2021, 9, 9846–9853. [Google Scholar] [CrossRef]
- Nunes, C.S.; da Silva, M.J.V.; da Silva, D.C.; dos R. Freitas, A.; Rosa, F.A.; Rubira, A.F.; Muniz, E.C. PET Depolymerisation in Supercritical Ethanol Catalysed by [Bmim][BF4]. RSC Adv. 2014, 4, 20308–20316. [Google Scholar] [CrossRef]
- Imran, M.; Kim, B.-K.; Han, M.; Cho, B.G.; Kim, D.H. Sub- and Supercritical Glycolysis of Polyethylene Terephthalate (PET) into the Monomer Bis(2-Hydroxyethyl) Terephthalate (BHET). Polym. Degrad. Stab. 2010, 95, 1686–1693. [Google Scholar] [CrossRef]
- Barboza, E.S.; Lopez, D.R.; Amico, S.C.; Ferreira, C.A. Determination of a Recyclability Index for the PET Glycolysis. Resour. Conserv. Recycl. 2009, 53, 122–128. [Google Scholar] [CrossRef]
- Ding, J.; Chen, J.; Ji, Y.; Ni, P.; Li, Z.; Xing, L. Kinetics of Alcoholysis of Poly(Ethylene Terephthalate) in Sub- and Super-Critical Isooctyl Alcohol to Produce Dioctyl Terephthalate. J. Anal. Appl. Pyrolysis 2014, 106, 99–103. [Google Scholar] [CrossRef]
- Liu, F.; Chen, J.; Li, Z.; Ni, P.; Ji, Y.; Meng, Q. Alcoholysis of Poly(Ethylene Terephthalate) to Produce Dioctyl Terephthalate with Sub- and Super-Critical Isooctyl Alcohol. J. Anal. Appl. Pyrolysis 2013, 99, 16–22. [Google Scholar] [CrossRef]
- Liu, J.; Qin, Z.; Wang, G.; Hou, X.; Wang, J. Critical Properties of Binary and Ternary Mixtures of Hexane+ Methanol, Hexane+ Carbon Dioxide, Methanol+ Carbon Dioxide, and Hexane+ Carbon Dioxide+ Methanol. J. Chem. Eng. Data 2003, 48, 1610–1613. [Google Scholar] [CrossRef]
- Pöhler, H.; Kiran, E. Volumetric Properties of Carbon Dioxide+ Ethanol at High Pressures. J. Chem. Eng. Data 1997, 42, 384–388. [Google Scholar] [CrossRef]
- Liu, J.; Yin, J. Carbon Dioxide Synergistic Enhancement of Supercritical Methanol on PET Depolymerization for Chemical Recovery. Ind. Eng. Chem. Res. 2022, 61, 6813–6819. [Google Scholar] [CrossRef]
- Yu, K.; Liu, J.; Sun, J.; Shen, Z.; Yin, J. Study of Polyester Degradation by Sub/Supercritical Ethanol and Enhancement of Carbon Dioxide. J. Supercrit. Fluids 2023, 194, 105837. [Google Scholar] [CrossRef]
- Li, X.-K.; Lu, H.; Guo, W.-Z.; Cao, G.-P.; Liu, H.-L.; Shi, Y.-H. Reaction Kinetics and Mechanism of Catalyzed Hydrolysis of Waste PET Using Solid Acid Catalyst in Supercritical CO2. AIChE J. 2015, 61, 200–214. [Google Scholar] [CrossRef]
- Guo, W.-Z.; Lu, H.; Li, X.-K.; Cao, G.-P. Tungsten-Promoted Titania as Solid Acid for Catalytic Hydrolysis of Waste Bottle PET in Supercritical CO2. RSC Adv. 2016, 6, 43171–43184. [Google Scholar] [CrossRef]
- Spyriouni, T.; Boulougouris, G.C.; Theodorou, D.N. Prediction of Sorption of CO2 in Glassy Atactic Polystyrene at Elevated Pressures through a New Computational Scheme. Macromolecules 2009, 42, 1759–1769. [Google Scholar] [CrossRef]
- Bozorgmehr, M.R.; Morsali, A.; Beyramabadi, S.A.; Moghaddam, F.K.; Pashirepour, J.; Shakeri, M. All Atom Molecular Dynamics Simulation Study of Polyethylene Polymer in Supercritical Water, Supercritical Ethanol and Supercritical Methanol. J. Supercrit. Fluids 2014, 86, 124–128. [Google Scholar] [CrossRef]
- Sun, F.; Dedong, H.; Fei, L.; Weiqiang, W.; Zhaotao, G.; Zhuo, Z. Molecular-Level Investigation of Plasticization of Polyethylene Terephthalate (PET) in Supercritical Carbon Dioxide via Molecular Dynamics Simulation. R. Soc. Open Sci. 2022, 9, 220606. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Yang, Y.; Shen, T.; Grisdanurak, N.; Pariatamby, A.; Khalid, M.; Hantoko, D.; Wibowo, H. Effect of Operating Parameters on Monomer Production from Depolymerization of Waste Polyethylene Terephthalate in Supercritical Ethanol. Process Saf. Environ. Prot. 2023, 169, 212–219. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Potoff, J.J.; Siepmann, J.I. Vapor–Liquid Equilibria of Mixtures Containing Alkanes, Carbon Dioxide, and Nitrogen. AIChE J. 2001, 47, 1676–1682. [Google Scholar] [CrossRef]
- Chen, B.; Potoff, J.J.; Siepmann, J.I. Monte Carlo Calculations for Alcohols and Their Mixtures with Alkanes. Transferable Potentials for Phase Equilibria. 5. United-Atom Description of Primary, Secondary, and Tertiary Alcohols. J. Phys. Chem. B 2001, 105, 3093–3104. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Tian Lu Sobtop Version 3.1. Available online: http://sobereva.com/soft/Sobtop/ (accessed on 25 July 2022).
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Berthelot, D. Sur Le Mélange Des Gaz. C. R. 1898, 126. Available online: https://www.biodiversitylibrary.org/item/23756#page/1/mode/1up (accessed on 1 April 2024).
- Lorentz, H.A. Ueber Die Anwendung Des Satzes Vom Virial in Der Kinetischen Theorie Der Gase. Ann. Der Phys. 1881, 248, 127–136. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Schmidt, J.R.; Skinner, J.L. Hydrogen Bonding Definitions and Dynamics in Liquid Water. J. Chem. Phys. 2007, 126, 204107. [Google Scholar] [CrossRef] [PubMed]
- Asakuma, Y.; Yamamura, Y.; Nakagawa, K.; Maeda, K.; Fukui, K. Mechanism of Depolymerization Reaction of Polyethylene Terephthalate: Experimental and Theoretical Studies. J. Polym. Env. 2011, 19, 209–216. [Google Scholar] [CrossRef]
- Mehl, A.; Nascimento, F.P.; Falcão, P.W.; Pessoa, F.L.P.; Cardozo-Filho, L. Vapor-Liquid Equilibrium of Carbon Dioxide + Ethanol: Experimental Measurements with Acoustic Method and Thermodynamic Modeling. J. Thermodyn. 2011, 2011. [Google Scholar] [CrossRef]
- Chiu, H.-Y.; Lee, M.-J.; Lin, H. Vapor-liquid Phase Boundaries of Binary Mixtures of Carbon Dioxide with Ethanol and Acetone. J. Chem. Eng. Data 2008, 53, 2393–2402. [Google Scholar] [CrossRef]
- Joung, S.N.; Yoo, C.W.; Shin, H.Y.; Kim, S.Y.; Yoo, K.-P.; Lee, C.S.; Huh, W.S. Measurements and Correlation of High-Pressure VLE of Binary CO2–Alcohol Systems (Methanol, Ethanol, 2-Methoxyethanol and 2-Ethoxyethanol). Fluid Phase Equilibria 2001, 185, 219–230. [Google Scholar] [CrossRef]
- Liu, C.; Ling, Y.; Wang, Z.; Zheng, W.; Sun, W.; Zhao, L. Unveiling the Microenvironments between Ionic Liquids and Methanol for Alcoholysis of Poly(Ethylene Terephthalate). Chem. Eng. Sci. 2022, 247, 117024. [Google Scholar] [CrossRef]
- Kazarian, S.G.; Vincent, M.F.; Bright, F.V.; Liotta, C.L.; Eckert, C.A. Specific Intermolecular Interaction of Carbon Dioxide with Polymers. J. Am. Chem. Soc. 1996, 118, 1729–1736. [Google Scholar] [CrossRef]
- Drohmann, C.; Beckman, E.J. Phase Behavior of Polymers Containing Ether Groups in Carbon Dioxide. J. Supercrit. Fluids 2002, 22, 103–110. [Google Scholar] [CrossRef]
- Pardal, F.; Tersac, G. Comparative Reactivity of Glycols in PET Glycolysis. Polym. Degrad. Stab. 2006, 91, 2567–2578. [Google Scholar] [CrossRef]
- Aida, T.; Aizawa, T.; Kanakubo, M.; Nanjo, H. Relation between Volume Expansion and Hydrogen Bond Networks for CO2-alcohol Mixtures at 40 °C. J. Phys. Chem. B 2010, 114, 13628–13636. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Tsuge, N.; Nagahama, K. Solubilities of Ethanol, 1-Propanol, 2-Propanol and 1- Butanol in Supercritical Carbon Dioxide at 313 K and 333 K. Fluid Phase Equilibria 1991, 67, 213–226. [Google Scholar] [CrossRef]
- Zheng, W.; Liu, C.; Wei, X.; Sun, W.; Zhao, L. Molecular-Level Swelling Behaviors of Poly (Ethylene Terephthalate) Glycolysis Using Ionic Liquids as Catalyst. Chem. Eng. Sci. 2023, 267, 118329. [Google Scholar] [CrossRef]
- Miller-Chou, B.A.; Koenig, J.L. A Review of Polymer Dissolution. Prog. Polym. Sci. 2003, 28, 1223–1270. [Google Scholar] [CrossRef]
- Gurina, D.L.; Budkov, Y.A.; Kiselev, M.G. Polylactide Nanoparticle Impregnation with Carbamazepine in Supercritical Media and Its Subsequent Release in Liquid Solvents: Insights from Molecular Simulation. J. Mol. Liq. 2022, 352, 118758. [Google Scholar] [CrossRef]
- Hirogaki, K.; Tabata, I.; Hisada, K.; Hori, T. An Investigation of the Interaction of Supercritical Carbon Dioxide with Poly(Ethylene Terephthalate) and the Effects of Some Additive Modifiers on the Interaction. J. Supercrit. Fluids 2005, 36, 166–172. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Number of PET Chain | Composition | Molar Ratio (CO2/Alcohol) |
|---|---|---|---|
| 1 | 6 | methanol | 0 |
| 2 | 6 | CO2 + methanol | 0.5 |
| 3 | 6 | ethanol | 0 |
| 4 | 6 | CO2 + ethanol | 0.5 |
| 5 | 6 | CO2 + ethanol | 1 |
| 6 | 6 | CO2 + ethanol | 1.5 |
| 7 | 6 | 1-propanol | 0 |
| 8 | 6 | CO2 + 1propanol | 0.5 |
| 9 | 6 | 1-butanol | 0 |
| 10 | 6 | CO2 + 1-butanol | 0.5 |
| 11 | 6 | 1-pentanol | 0 |
| 12 | 6 | CO2 + 1pentanol | 0.5 |
| Reaction Time (min) | Mn [g/mol] (NCO2:Nethanol = 0) | Mn [g/mol] (NCO2:Nethanol = 0.2) |
|---|---|---|
| 0 | 12,586 | 12,586 |
| 20 | 10,083 | 10,227 |
| 30 | 9791 | 1143 |
| 40 | 5796 | 750 |
| 50 | 2771 | 580 |
| 60 | 1889 | 546 |
| HBs Donor-Accepter | NCO2:Nethanol | ||||
|---|---|---|---|---|---|
| 0 | 0.5 | 1.0 | 1.5 | ||
| Average Number of HBs | ethanol-PET | 197.825 | 178.834 | 135.765 | 113.363 |
| ethanol-CO2 | 0 | 14.504 | 26.180 | 26.207 | |
| Average Lifetime of HBs [ps] | ethanol-PET | 1.54 | 1.49 | 1.5 | 1.5 |
| ethanol-CO2 | 0 | 0.54 | 0.53 | 0.52 | |
| NCO2:Nethanol | Type of Alcohol Molecules | |||||
|---|---|---|---|---|---|---|
| Methanol | Ethanol | 1-Propanol | 1-Butanol | 1-Pentanol | ||
| Average Number of HBs | 0 | 201.76 | 197.825 | 190.883 | 186.988 | 186.295 |
| 0.5 | 184.735 | 178.834 | 171.083 | 164.405 | 159.688 | |
| Average Lifetime of HBs [ps] | 0 | 1.41 | 1.54 | 1.57 | 1.59 | 1.61 |
| 0.5 | 1.4 | 1.49 | 1.52 | 1.54 | 1.57 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Cui, R.; Han, Y.; Jiang, J.; Hu, D.; Zhao, L.; Xi, Z. Efficient Alcoholysis of Poly(ethylene terephthalate) by Using Supercritical Carbon Dioxide as a Green Solvent. Polymers 2024, 16, 1564. https://doi.org/10.3390/polym16111564
Xu Y, Cui R, Han Y, Jiang J, Hu D, Zhao L, Xi Z. Efficient Alcoholysis of Poly(ethylene terephthalate) by Using Supercritical Carbon Dioxide as a Green Solvent. Polymers. 2024; 16(11):1564. https://doi.org/10.3390/polym16111564
Chicago/Turabian StyleXu, Yiwei, Ran Cui, Yuqing Han, Jie Jiang, Dongdong Hu, Ling Zhao, and Zhenhao Xi. 2024. "Efficient Alcoholysis of Poly(ethylene terephthalate) by Using Supercritical Carbon Dioxide as a Green Solvent" Polymers 16, no. 11: 1564. https://doi.org/10.3390/polym16111564
APA StyleXu, Y., Cui, R., Han, Y., Jiang, J., Hu, D., Zhao, L., & Xi, Z. (2024). Efficient Alcoholysis of Poly(ethylene terephthalate) by Using Supercritical Carbon Dioxide as a Green Solvent. Polymers, 16(11), 1564. https://doi.org/10.3390/polym16111564