
Harnessing Data Augmentation and Normalization Preprocessing to Improve the Performance of Chemical Reaction Predictions of Data-Driven Model



Abstract
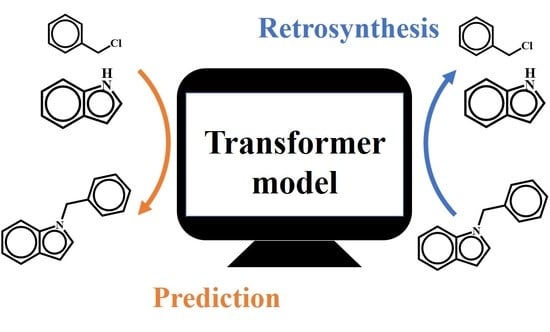
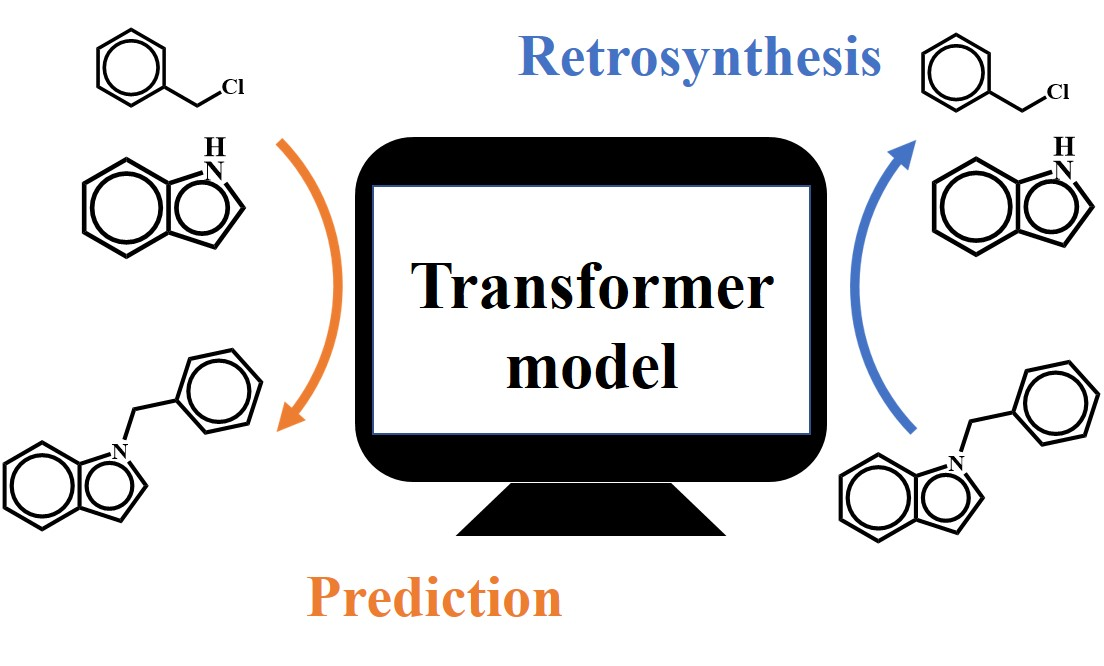{kind=link}
{kind=link}
{kind=link}
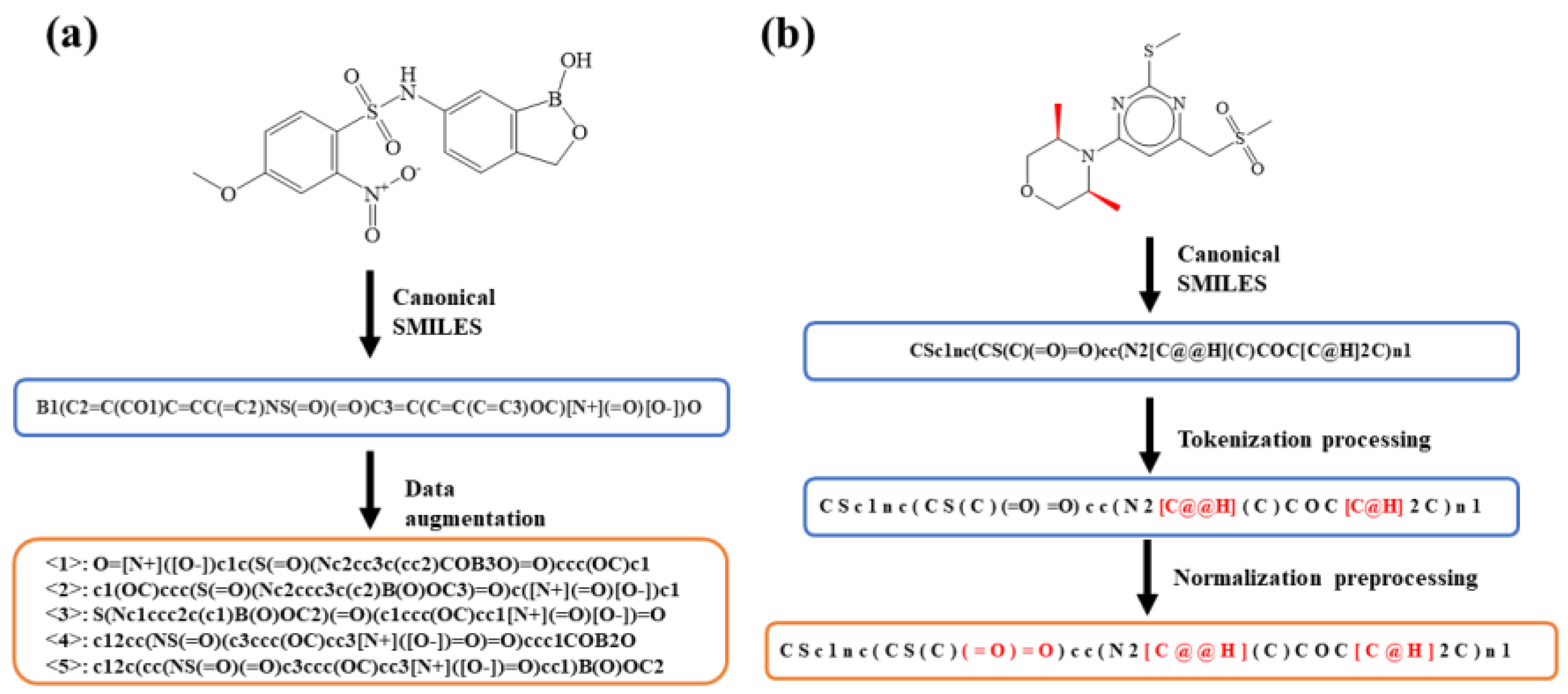{kind=link}
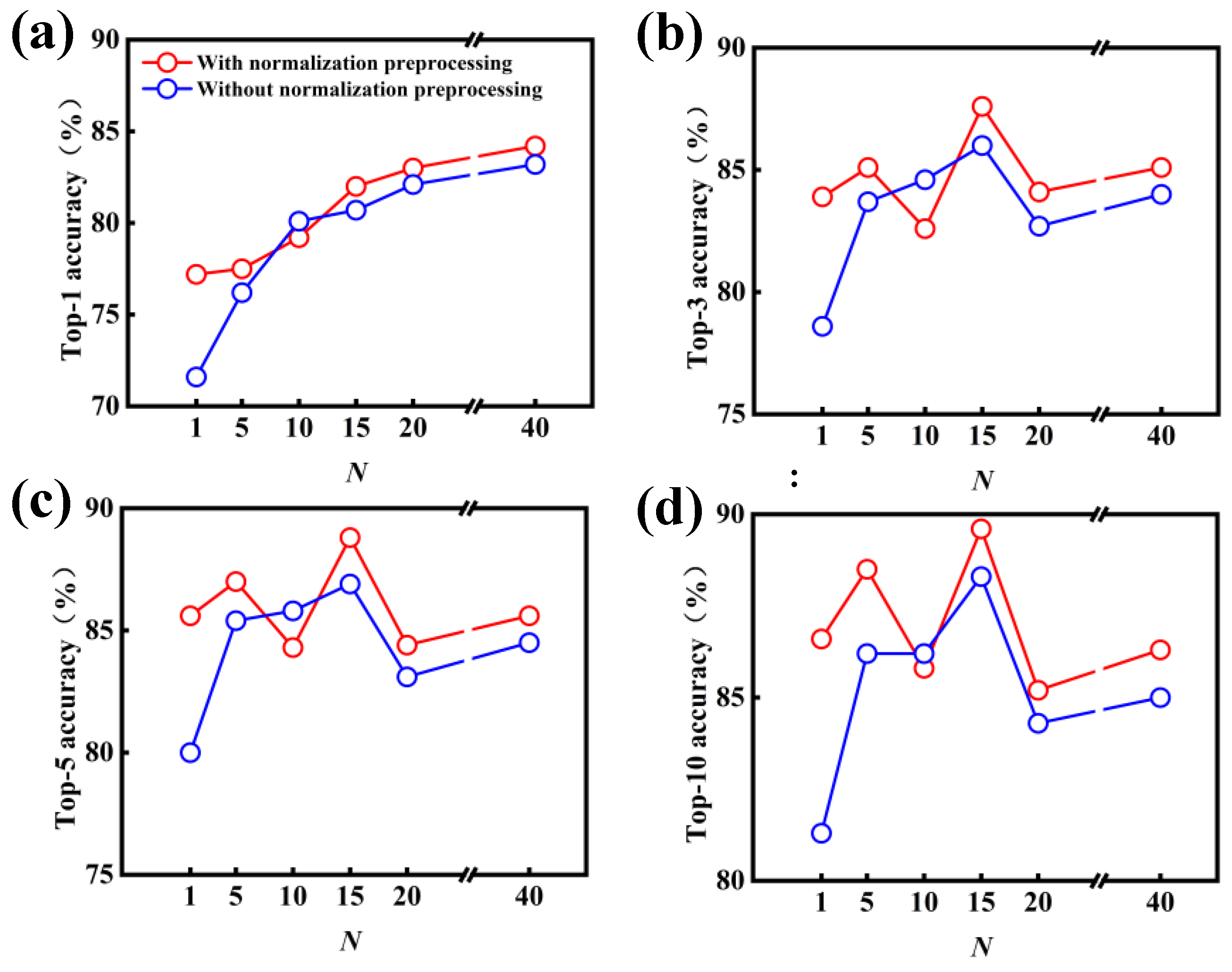{kind=link}
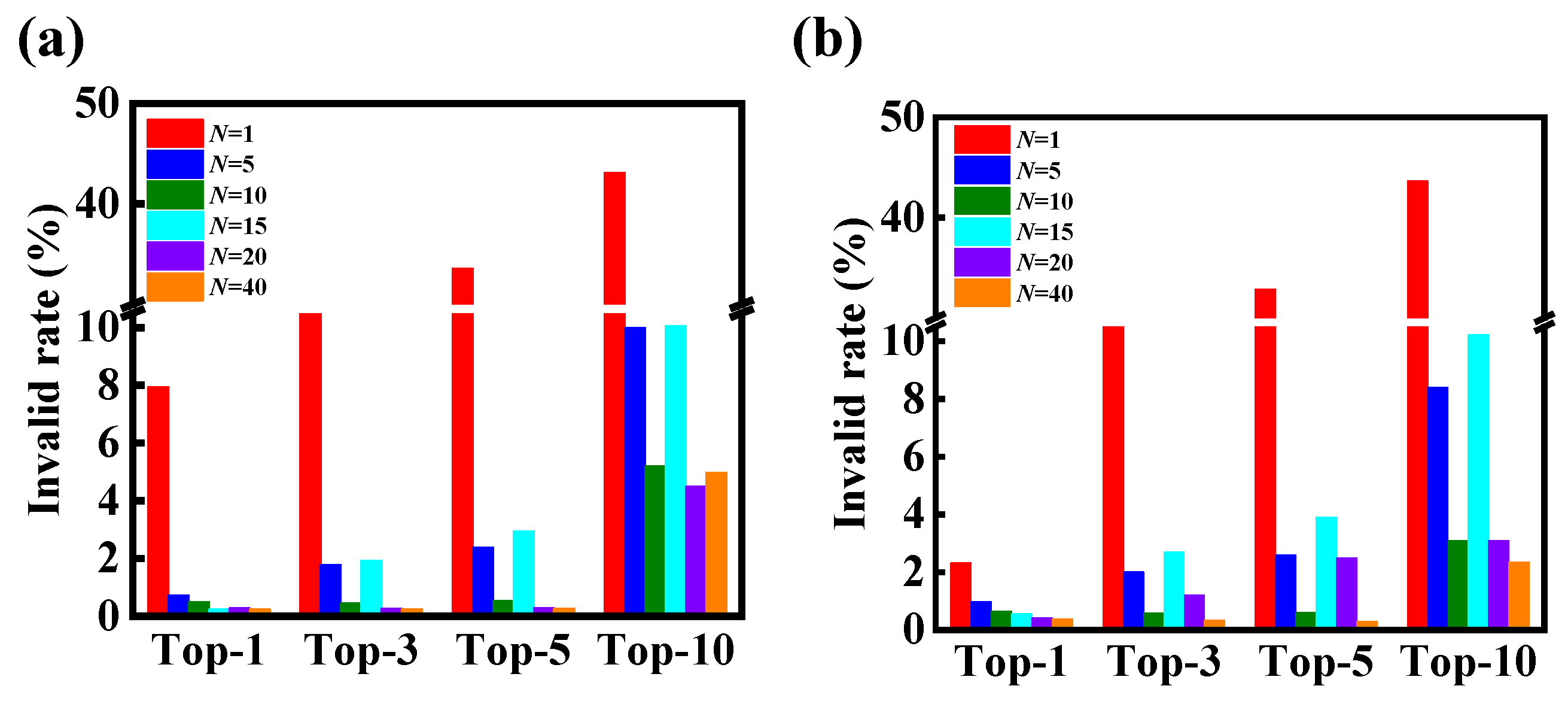{kind=link}
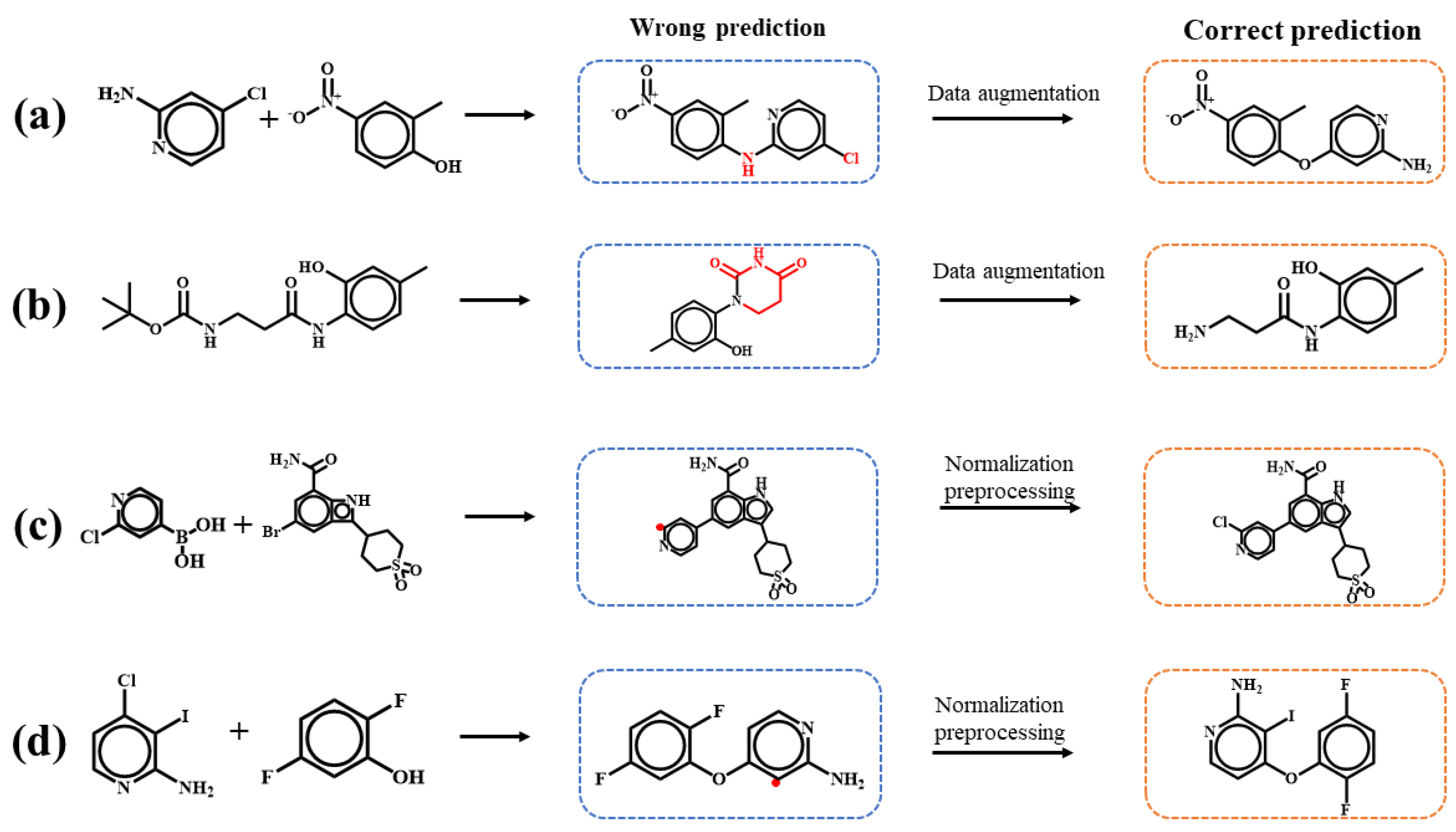{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Dataset and Methods
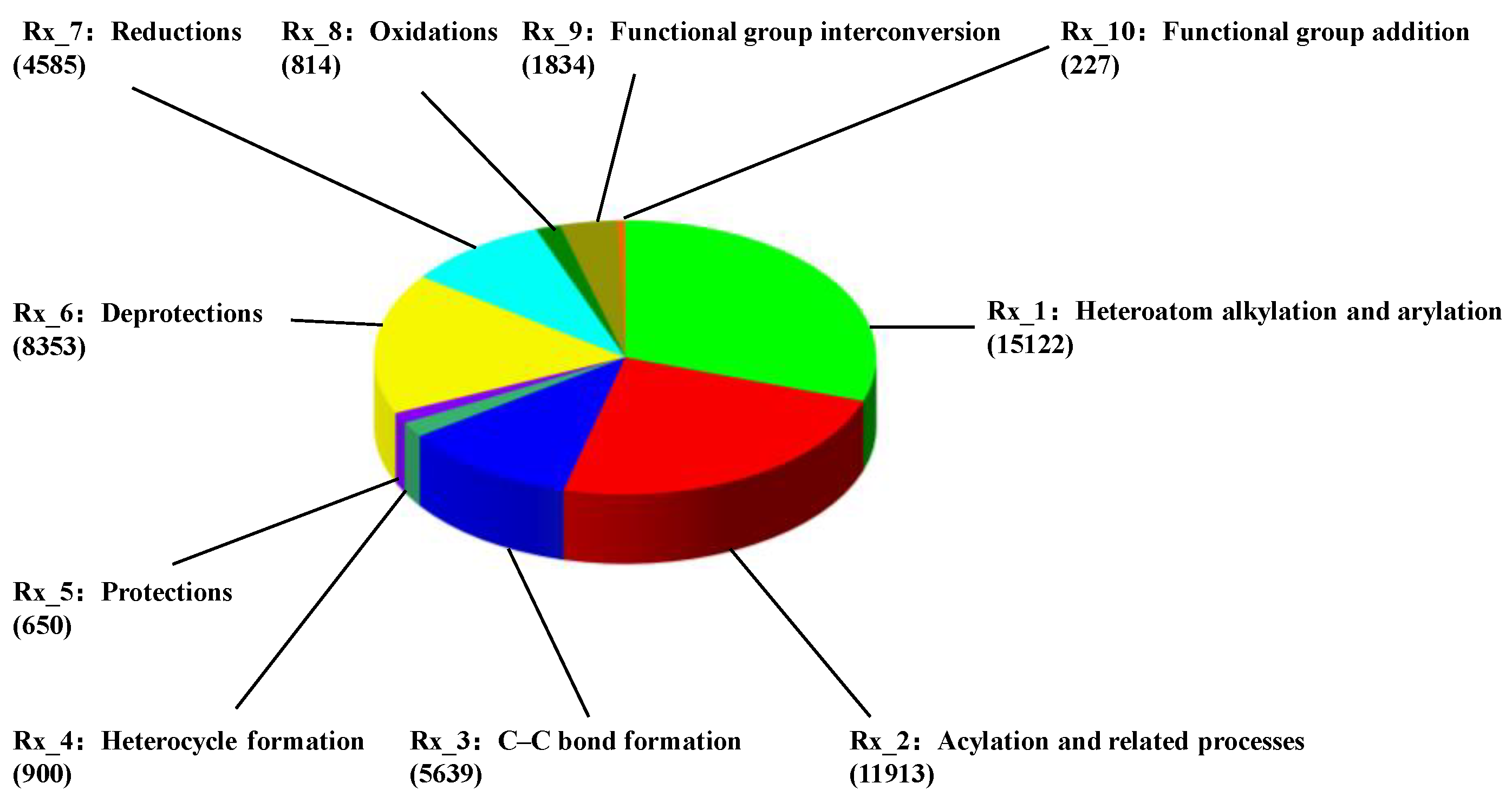
2.1. Dataset
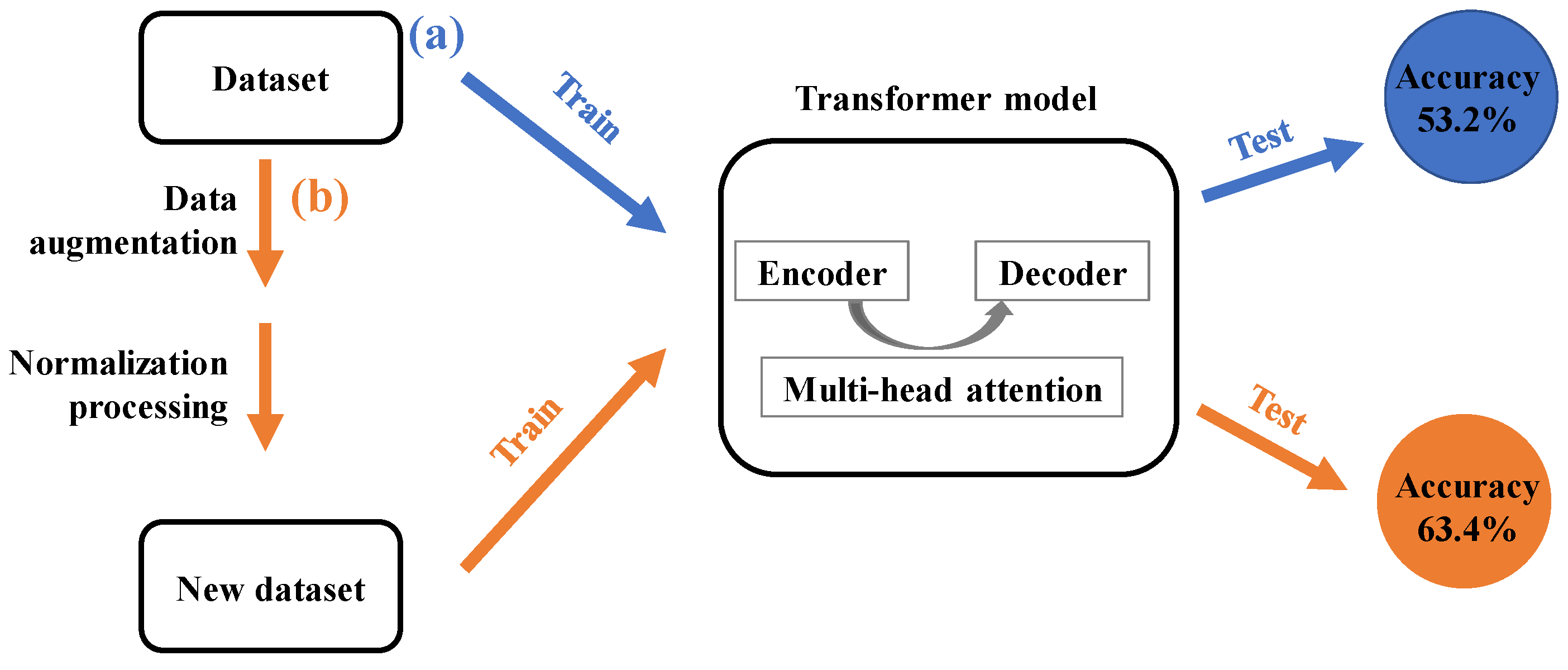
2.2. Data Preprocessing
2.3. Model
3. Results and Discussion
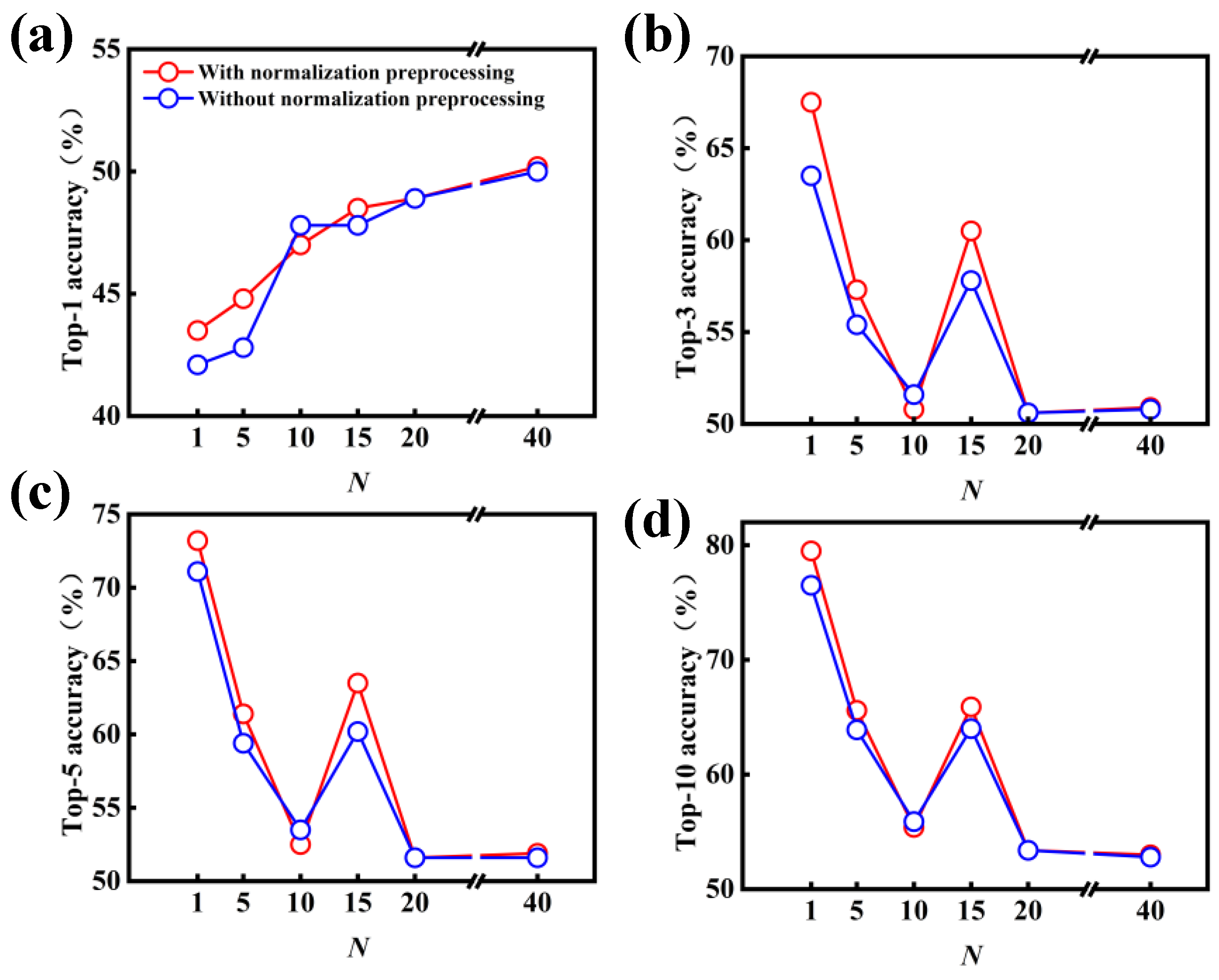
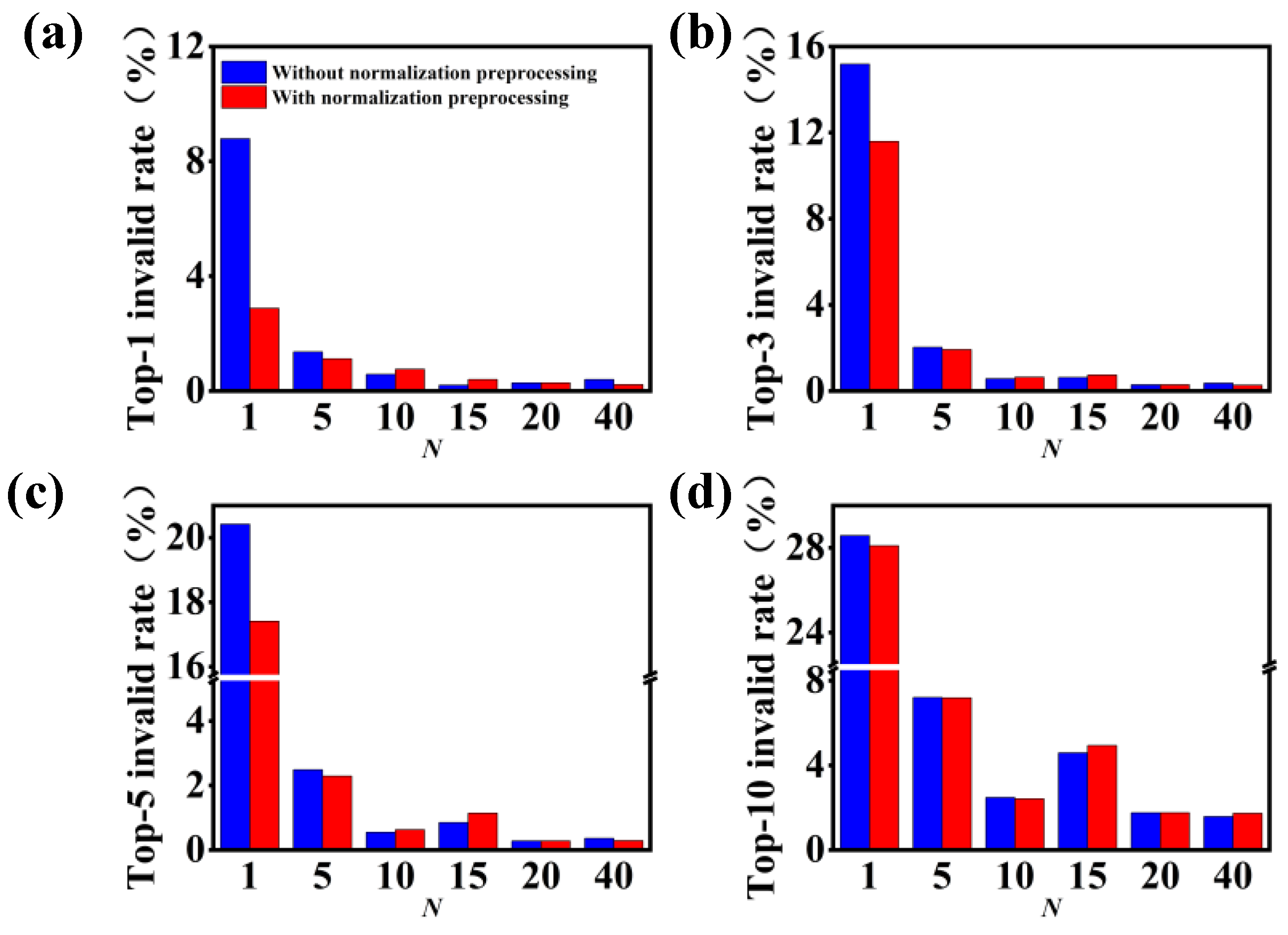
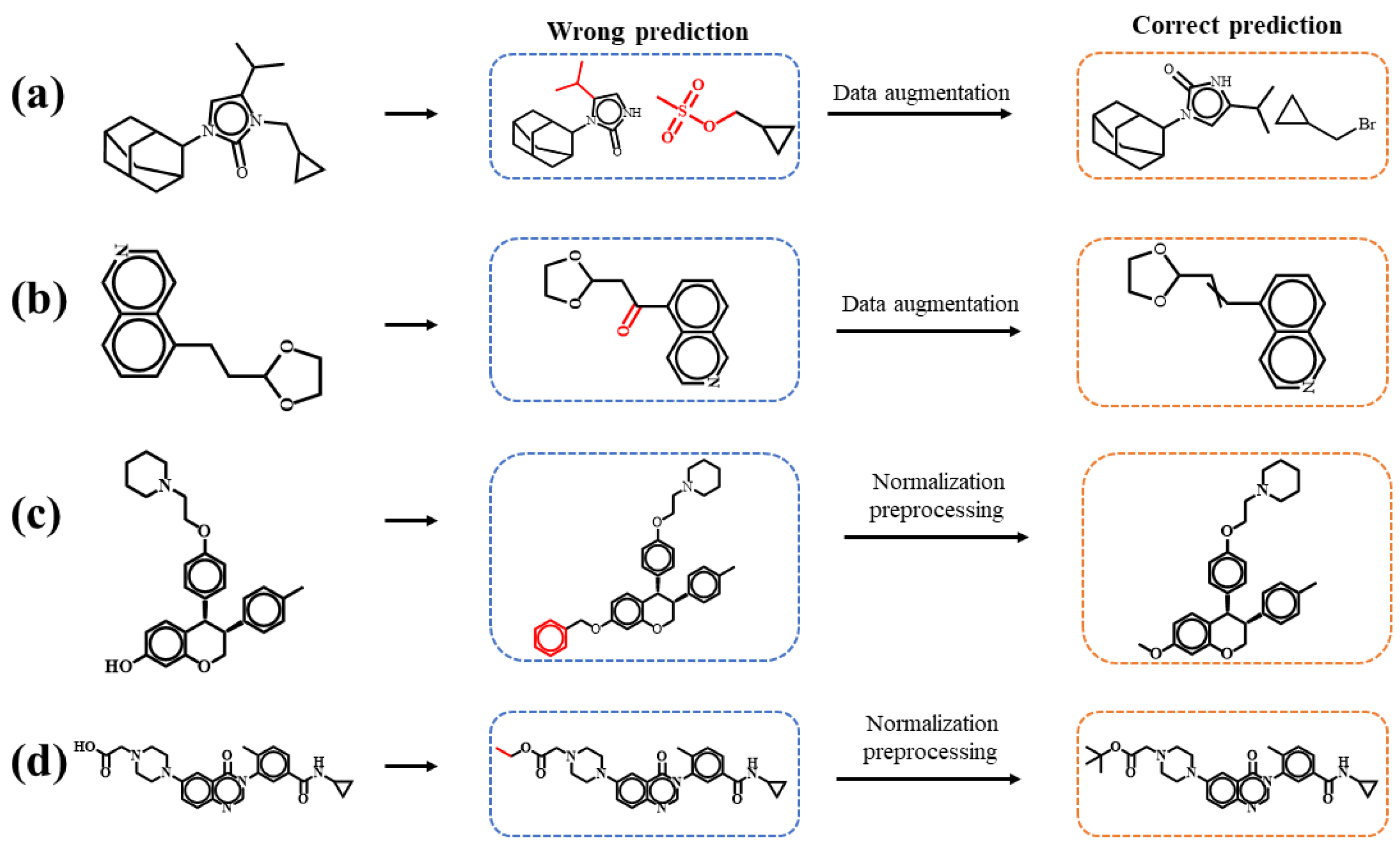
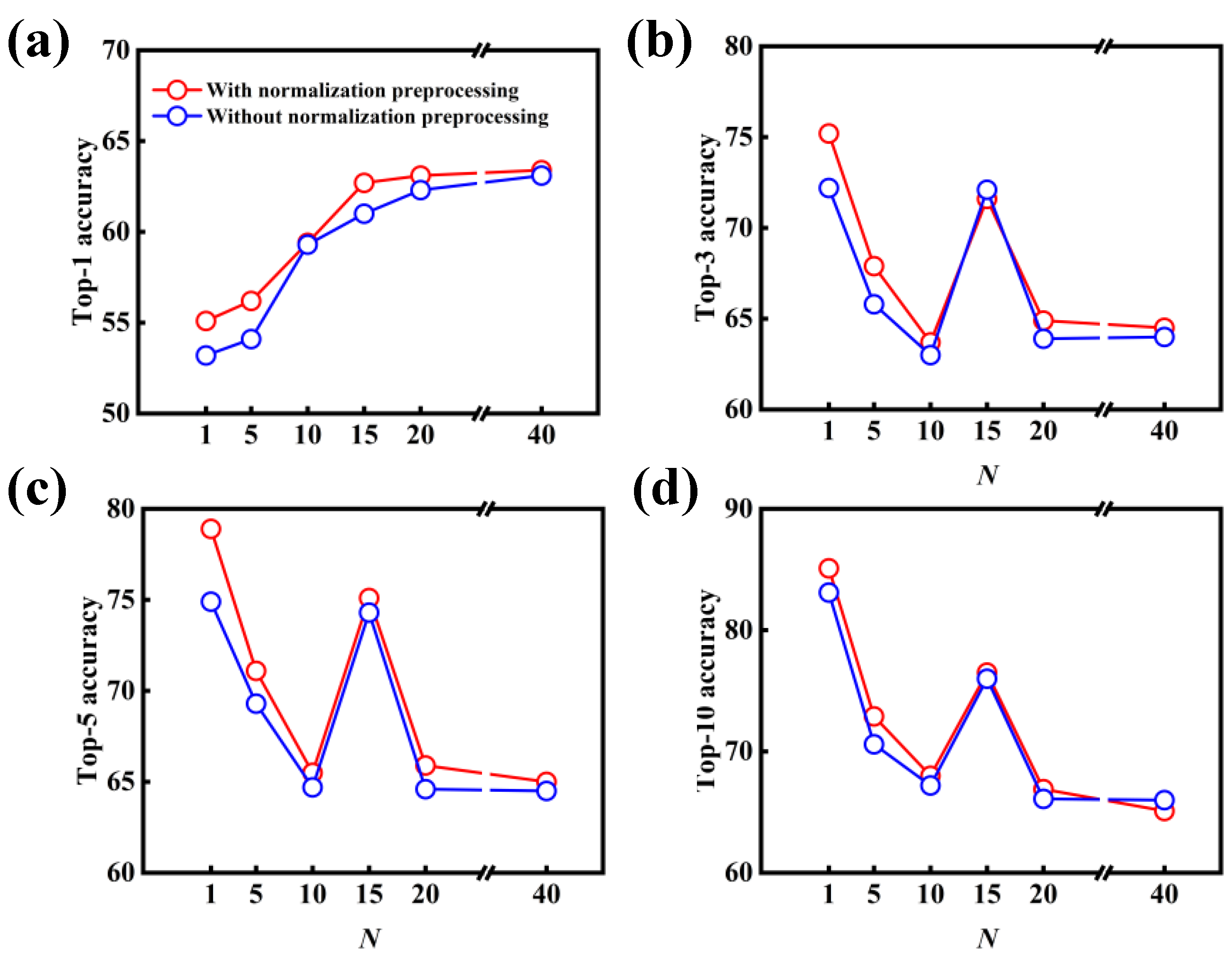
3.1. Model Performance on Forward Predictions of Chemical Reactions
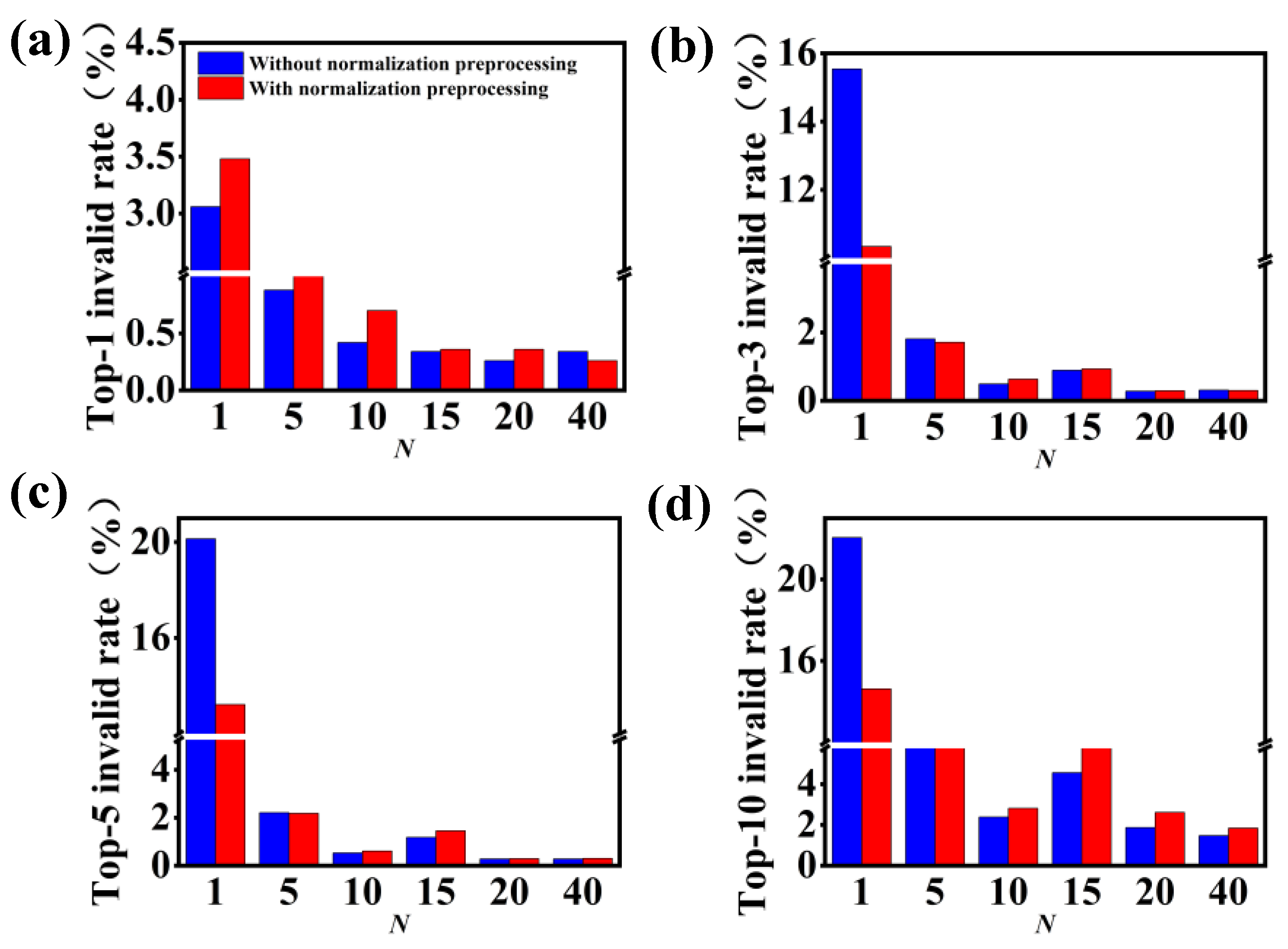
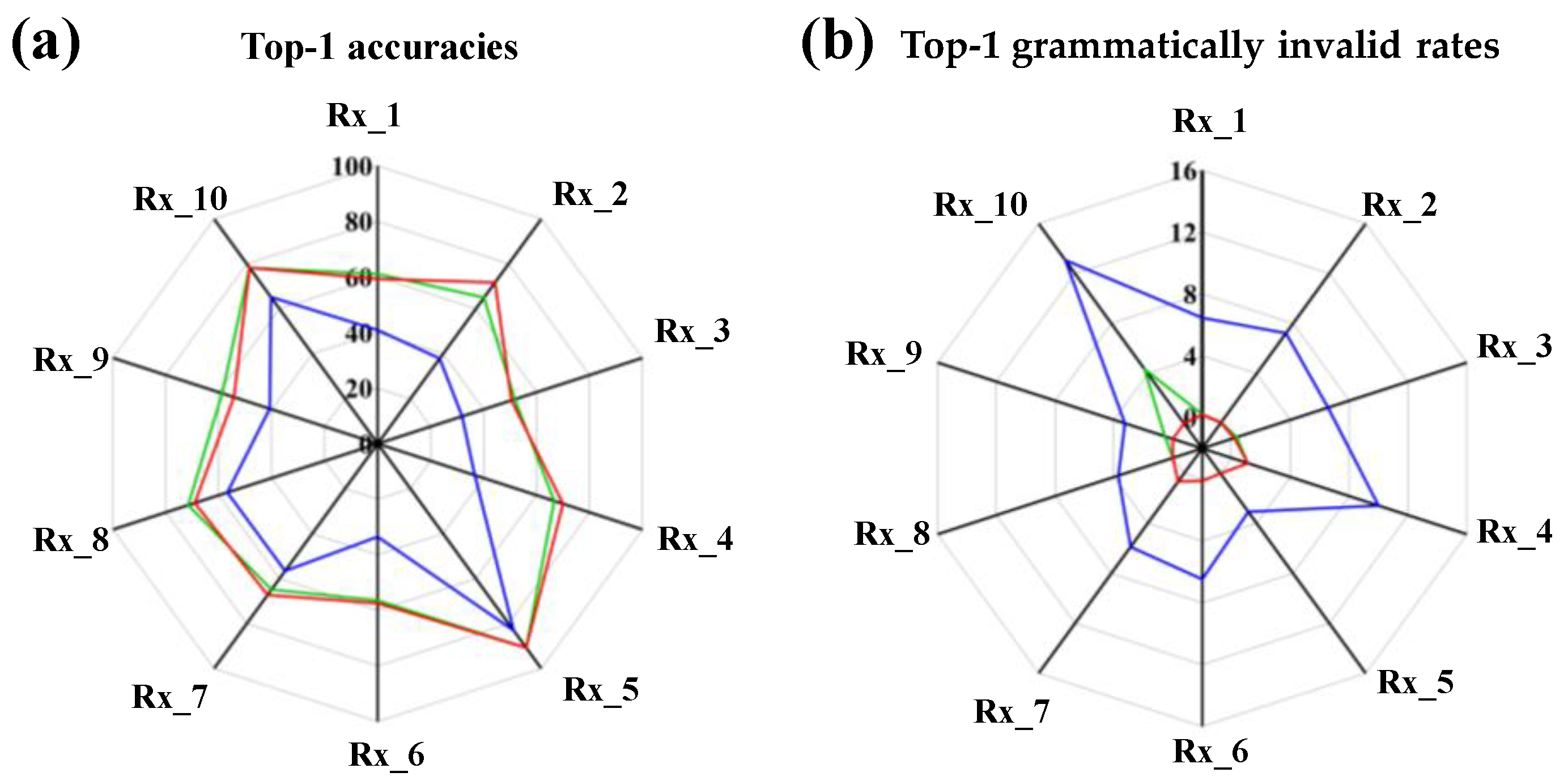
3.2. Model Performance on Single-Step Retrosynthesis without Reaction Classes
3.3. Model Performance on Single-Step Retrosynthesis with Reaction Classes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Corey, E.J.; Wipke, W.T. Computer-assisted design of complex organic syntheses: Pathways for molecular synthesis can be devised with a computer and equipment for graphical communication. Science 1969, 166, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, D.C.; Castro, L.; Churcher, I.; Rees, D.C.; Thomas, A.W.; Wilson, D.M.; Wood, A. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 2018, 10, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Segler, M.H.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic ai. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef]
- Collins, K.D.; Glorius, F. A robustness screen for the rapid assessment of chemical reactions. Nat. Chem. 2013, 5, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Szymkuć, S.; Gajewska, E.P.; Klucznik, T.; Molga, K.; Dittwald, P.; Startek, M.; Bajczyk, M.; Grzybowski, B.A. Computer-assisted synthetic planning: The end of the beginning. Angew. Chem. Int. Ed. 2016, 55, 5904–5937. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Long, A.K.; Rubenstein, S.D. Computer-assisted analysis in organic synthesis. Science 1985, 228, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.J.; Klajn, R.; Grzybowski, B.A. The core and most useful molecules in organic chemistry. Angew. Chem. Int. Ed. 2006, 45, 5348–5354. [Google Scholar] [CrossRef]
- Kowalik, M.; Gothard, C.M.; Drews, A.M.; Gothard, N.A.; Weckiewicz, A.; Fuller, P.E.; Grzybowski, B.A.; Bishop, K.J. Parallel optimization of synthetic pathways within the network of organic chemistry. Angew. Chem. Int. Ed. 2012, 51, 7928–7932. [Google Scholar] [CrossRef]
- Badowski, T.; Molga, K.; Grzybowski, B.A. Selection of cost-effective yet chemically diverse pathways from the networks of computer-generated retrosynthetic plans. Chem. Sci. 2019, 10, 4640–4651. [Google Scholar] [CrossRef]
- Mikulak-Klucznik, B.; Gołębiowska, P.; Bayly, A.A.; Popik, O.; Klucznik, T.; Szymkuć, S.; Gajewska, E.P.; Dittwald, P.; Staszewska-Krajewska, O.; Beker, W. Computational planning of the synthesis of complex natural products. Nature 2020, 588, 83–88. [Google Scholar] [CrossRef]
- Gothard, C.M.; Soh, S.; Gothard, N.A.; Kowalczyk, B.; Wei, Y.; Baytekin, B.; Grzybowski, B.A. Rewiring chemistry: Algorithmic discovery and experimental validation of one-pot reactions in the network of organic chemistry. Angew. Chem. 2012, 124, 8046–8051. [Google Scholar] [CrossRef]
- Martínez, T.J. Ab initio reactive computer aided molecular design. Acc. Chem. Res. 2017, 50, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-P.; Titov, A.; McGibbon, R.; Liu, F.; Pande, V.S.; Martínez, T.J. Discovering chemistry with an ab initio nanoreactor. Nat. Chem. 2014, 6, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Helma, C.; Cramer, T.; Kramer, S.; De Raedt, L. Data mining and machine learning techniques for the identification of mutagenicity inducing substructures and structure activity relationships of noncongeneric compounds. J. Chem. Inf. Comput. Sci. 2004, 44, 1402–1411. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Duan, J.; Smith, E.; Von Bargen, C.D.; Sherman, W.; Repasky, M.P. Autoqsar: An automated machine learning tool for best-practice quantitative structure–activity relationship modeling. Future Med. Chem. 2016, 8, 1825–1839. [Google Scholar] [CrossRef] [PubMed]
- King, R.D.; Muggleton, S.; Lewis, R.A.; Sternberg, M. Drug design by machine learning: The use of inductive logic programming to model the structure-activity relationships of trimethoprim analogues binding to dihydrofolate reductase. Proc. Natl. Acad. Sci. USA 1992, 89, 11322–11326. [Google Scholar] [CrossRef]
- Xiao, Z.; Yang, B.; Feng, X.; Liao, Z.; Shi, H.; Jiang, W.; Wang, C.; Ren, N. Density functional theory and machine learning-based quantitative structure–activity relationship models enabling prediction of contaminant degradation performance with heterogeneous peroxymonosulfate treatments. Environ. Sci. Technol. 2023, 57, 3951–3961. [Google Scholar] [CrossRef]
- Ain, Q.U.; Aleksandrova, A.; Roessler, F.D.; Ballester, P.J. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 405–424. [Google Scholar] [CrossRef]
- Melville, J.L.; Burke, E.K.; Hirst, J.D. Machine learning in virtual screening. Comb. Chem. High Throughput Screen. 2009, 12, 332–343. [Google Scholar] [CrossRef]
- Axelrod, S.; Shakhnovich, E.; Gómez-Bombarelli, R. Thermal half-lives of azobenzene derivatives: Virtual screening based on intersystem crossing using a machine learning potential. ACS Cent. Sci. 2023, 9, 166–176. [Google Scholar] [CrossRef]
- Soleimany, A.P.; Amini, A.; Goldman, S.; Rus, D.; Bhatia, S.N.; Coley, C.W. Evidential deep learning for guided molecular property prediction and discovery. ACS Cent. Sci. 2021, 7, 1356–1367. [Google Scholar] [CrossRef] [PubMed]
- Jinich, A.; Sanchez-Lengeling, B.; Ren, H.; Harman, R.; Aspuru-Guzik, A. A mixed quantum chemistry/machine learning approach for the fast and accurate prediction of biochemical redox potentials and its large-scale application to 315,000 redox reactions. ACS Cent. Sci. 2019, 5, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Dral, P.O. Quantum chemistry in the age of machine learning. J. Phys. Chem. Lett. 2020, 11, 2336–2347. [Google Scholar] [CrossRef] [PubMed]
- Schütt, K.T.; Gastegger, M.; Tkatchenko, A.; Müller, K.-R.; Maurer, R.J. Unifying machine learning and quantum chemistry with a deep neural network for molecular wavefunctions. Nat. Commun. 2019, 10, 5024. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Kais, S. Quantum machine learning for electronic structure calculations. Nat. Commun. 2018, 9, 4195. [Google Scholar] [CrossRef]
- Chen, S.; Jung, Y. Deep retrosynthetic reaction prediction using local reactivity and global attention. JACS Au 2021, 1, 1612–1620. [Google Scholar] [CrossRef]
- Coley, C.W.; Jin, W.; Rogers, L.; Jamison, T.F.; Jaakkola, T.S.; Green, W.H.; Barzilay, R.; Jensen, K.F. A graph-convolutional neural network model for the prediction of chemical reactivity. Chem. Sci. 2019, 10, 370–377. [Google Scholar] [CrossRef]
- Nam, J.; Kim, J. Linking the neural machine translation and the prediction of organic chemistry reactions. arXiv 2016, arXiv:1612.09529. [Google Scholar]
- Wei, J.N.; Duvenaud, D.; Aspuru-Guzik, A. Neural networks for the prediction of organic chemistry reactions. ACS Cent. Sci. 2016, 2, 725–732. [Google Scholar] [CrossRef]
- Coley, C.W.; Barzilay, R.; Jaakkola, T.S.; Green, W.H.; Jensen, K.F. Prediction of organic reaction outcomes using machine learning. ACS Cent. Sci. 2017, 3, 434–443. [Google Scholar] [CrossRef]
- Liu, B.; Ramsundar, B.; Kawthekar, P.; Shi, J.; Gomes, J.; Luu Nguyen, Q.; Ho, S.; Sloane, J.; Wender, P.; Pande, V. Retrosynthetic reaction prediction using neural sequence-to-sequence models. ACS Cent. Sci. 2017, 3, 1103–1113. [Google Scholar] [CrossRef]
- Weininger, D. Smiles, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. In Advances in Neural Information Processing Systems; MIT Press: Cambridge, MA, USA, 2017; Volume 30. [Google Scholar]
- Schwaller, P.; Laino, T.; Gaudin, T.; Bolgar, P.; Hunter, C.A.; Bekas, C.; Lee, A.A. Molecular transformer: A model for uncertainty-calibrated chemical reaction prediction. ACS Cent. Sci. 2019, 5, 1572–1583. [Google Scholar] [CrossRef]
- Tetko, I.V.; Karpov, P.; Bruno, E.; Kimber, T.B.; Godin, G. Augmentation is what you need! In Artificial Neural Networks and Machine Learning—ICANN 2019: Workshop and Special Sessions, Proceedings of the 28th International Conference on Artificial Neural Networks, Munich, Germany, 17–19 September 2019; Springer: Berlin/Heidelberg, Germany, 2019; pp. 831–835. [Google Scholar]
- Fortunato, M.E.; Coley, C.W.; Barnes, B.C.; Jensen, K.F. Data augmentation and pretraining for template-based retrosynthetic prediction in computer-aided synthesis planning. J. Chem. Inf. Model. 2020, 60, 3398–3407. [Google Scholar] [CrossRef] [PubMed]
- Kimber, T.B.; Engelke, S.; Tetko, I.V.; Bruno, E.; Godin, G. Synergy effect between convolutional neural networks and the multiplicity of smiles for improvement of molecular prediction. arXiv 2018, arXiv:1812.04439. [Google Scholar]
- Lowe, D.M. Extraction of Chemical Structures and Reactions from the Literature. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 2012. [Google Scholar]
- Weininger, D.; Weininger, A.; Weininger, J.L. Smiles. 2. Algorithm for generation of unique smiles notation. J. Chem. Inf. Comput. Sci. 1989, 29, 97–101. [Google Scholar] [CrossRef]
- Karpov, P.; Godin, G.; Tetko, I.V. Transformer-cnn: Swiss knife for qsar modeling and interpretation. J. Cheminformatics 2020, 12, 17. [Google Scholar] [CrossRef]
- Jannik Bjerrum, E. Smiles enumeration as data augmentation for neural network modeling of molecules. arXiv 2017, arXiv:1703.07076. [Google Scholar]
- Schwaller, P.; Gaudin, T.; Lanyi, D.; Bekas, C.; Laino, T. “Found in translation”: Predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem. Sci. 2018, 9, 6091–6098. [Google Scholar] [CrossRef]
- Klein, G.; Kim, Y.; Deng, Y.; Senellart, J.; Rush, A.M. Opennmt: Open-source toolkit for neural machine translation. arXiv 2017, arXiv:1701.02810. [Google Scholar]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Lin, J.; Du, L.; Zhang, L. Harnessing Data Augmentation and Normalization Preprocessing to Improve the Performance of Chemical Reaction Predictions of Data-Driven Model. Polymers 2023, 15, 2224. https://doi.org/10.3390/polym15092224
Zhang B, Lin J, Du L, Zhang L. Harnessing Data Augmentation and Normalization Preprocessing to Improve the Performance of Chemical Reaction Predictions of Data-Driven Model. Polymers. 2023; 15(9):2224. https://doi.org/10.3390/polym15092224
Chicago/Turabian StyleZhang, Boyu, Jiaping Lin, Lei Du, and Liangshun Zhang. 2023. "Harnessing Data Augmentation and Normalization Preprocessing to Improve the Performance of Chemical Reaction Predictions of Data-Driven Model" Polymers 15, no. 9: 2224. https://doi.org/10.3390/polym15092224
APA StyleZhang, B., Lin, J., Du, L., & Zhang, L. (2023). Harnessing Data Augmentation and Normalization Preprocessing to Improve the Performance of Chemical Reaction Predictions of Data-Driven Model. Polymers, 15(9), 2224. https://doi.org/10.3390/polym15092224