
Graphene Oxide and Polymer Humidity Micro-Sensors Prepared by Carbon Beam Writing

 , ,
, ,  , ,
, , 
Abstract
1. Introduction
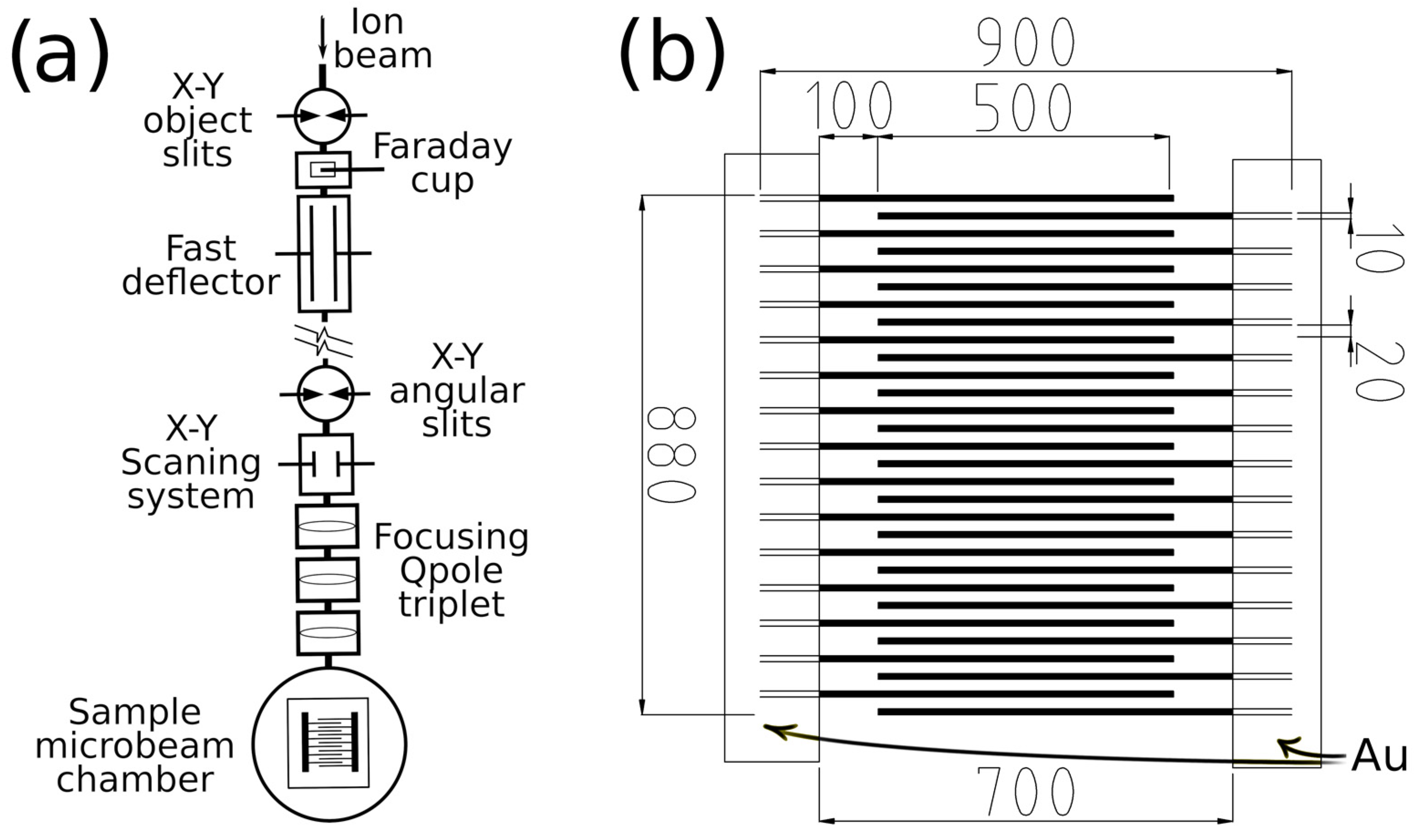
2. Materials and Methods
3. Results
3.1. Elemental Composition by RBS and ERDA
3.2. Surface Morphology by SEM
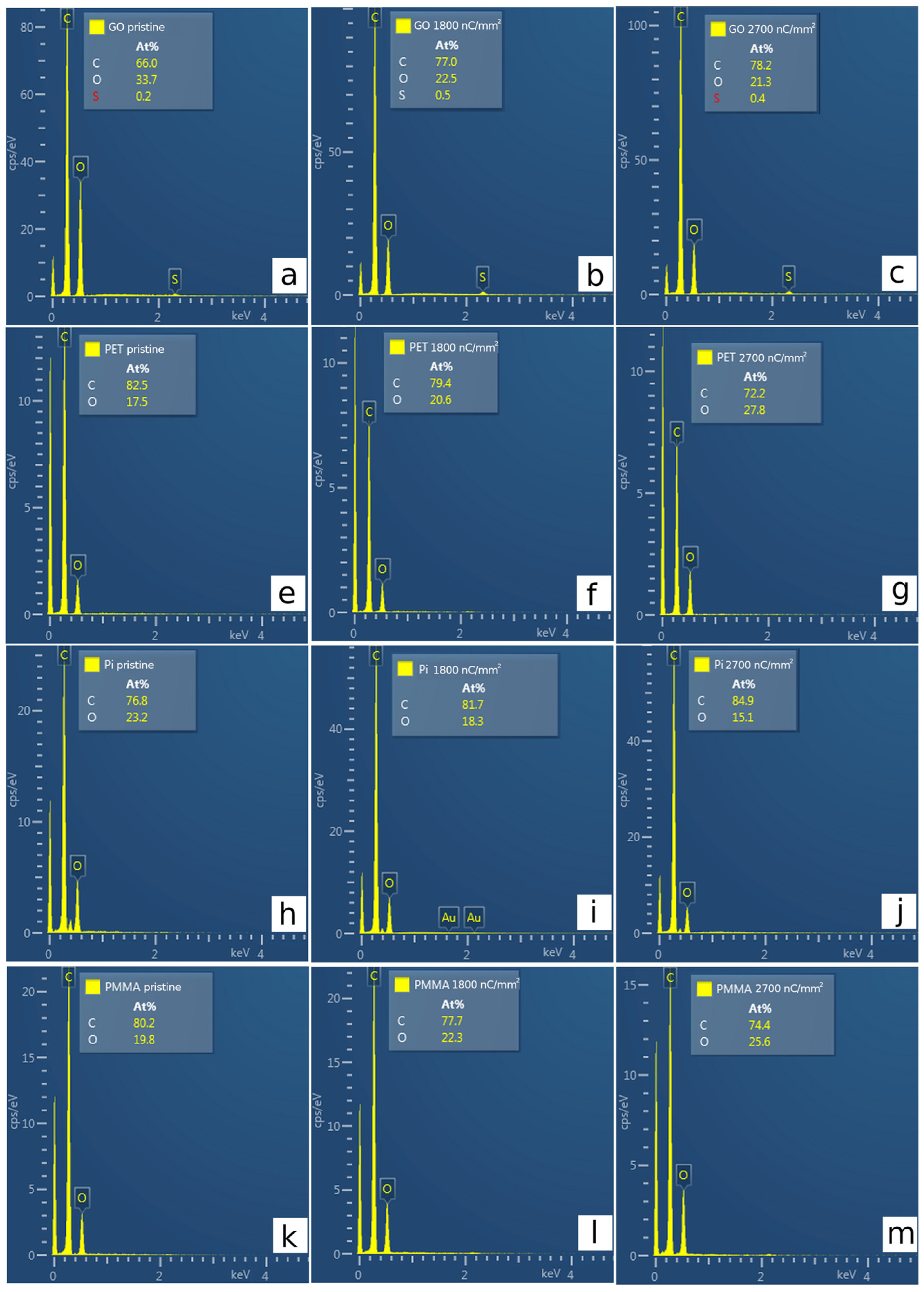
3.3. Elemental Composition by EDS
3.4. Structure Analysis by Raman Spectroscopy
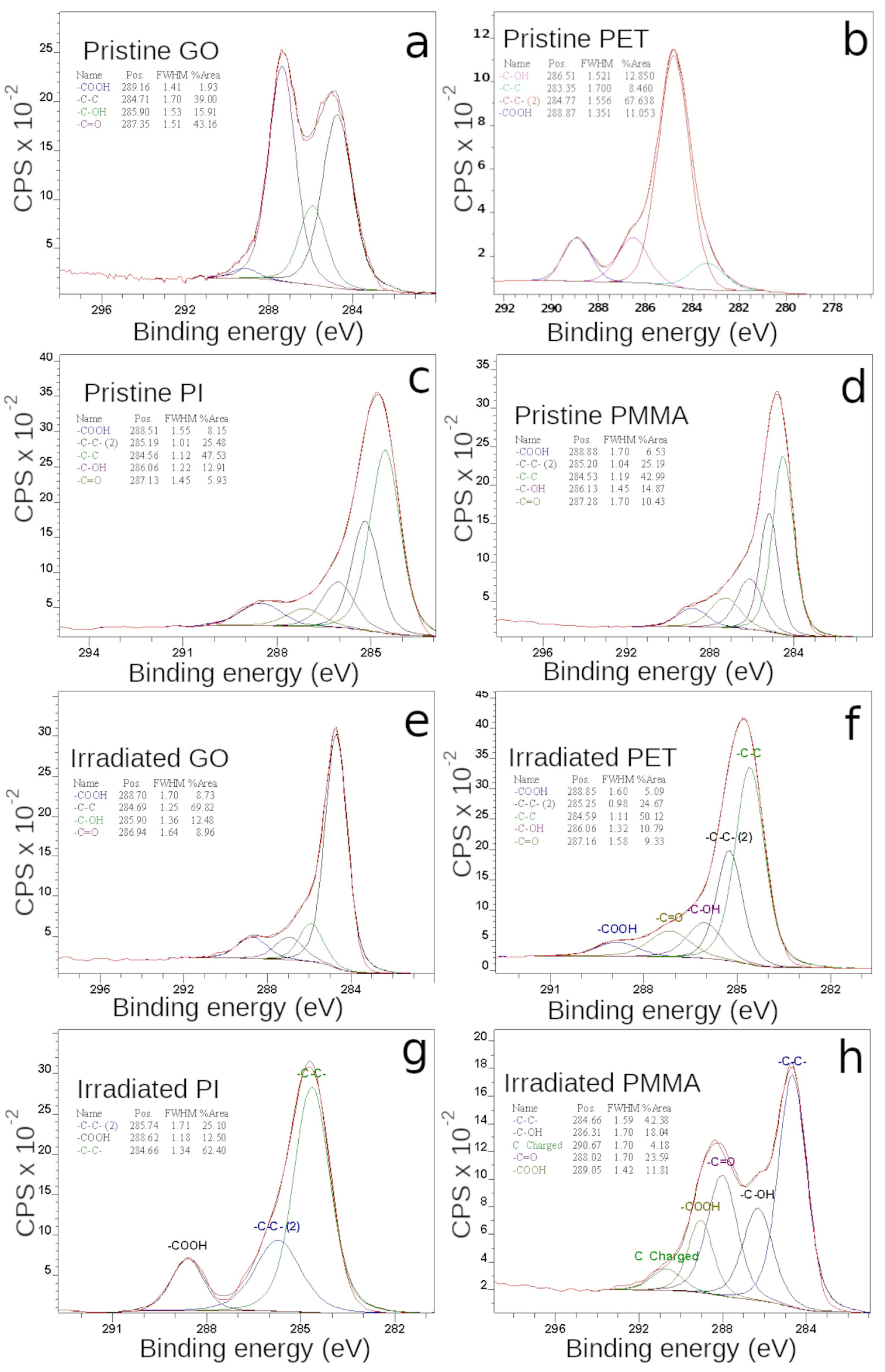
3.5. Surface Chemistry by XPS
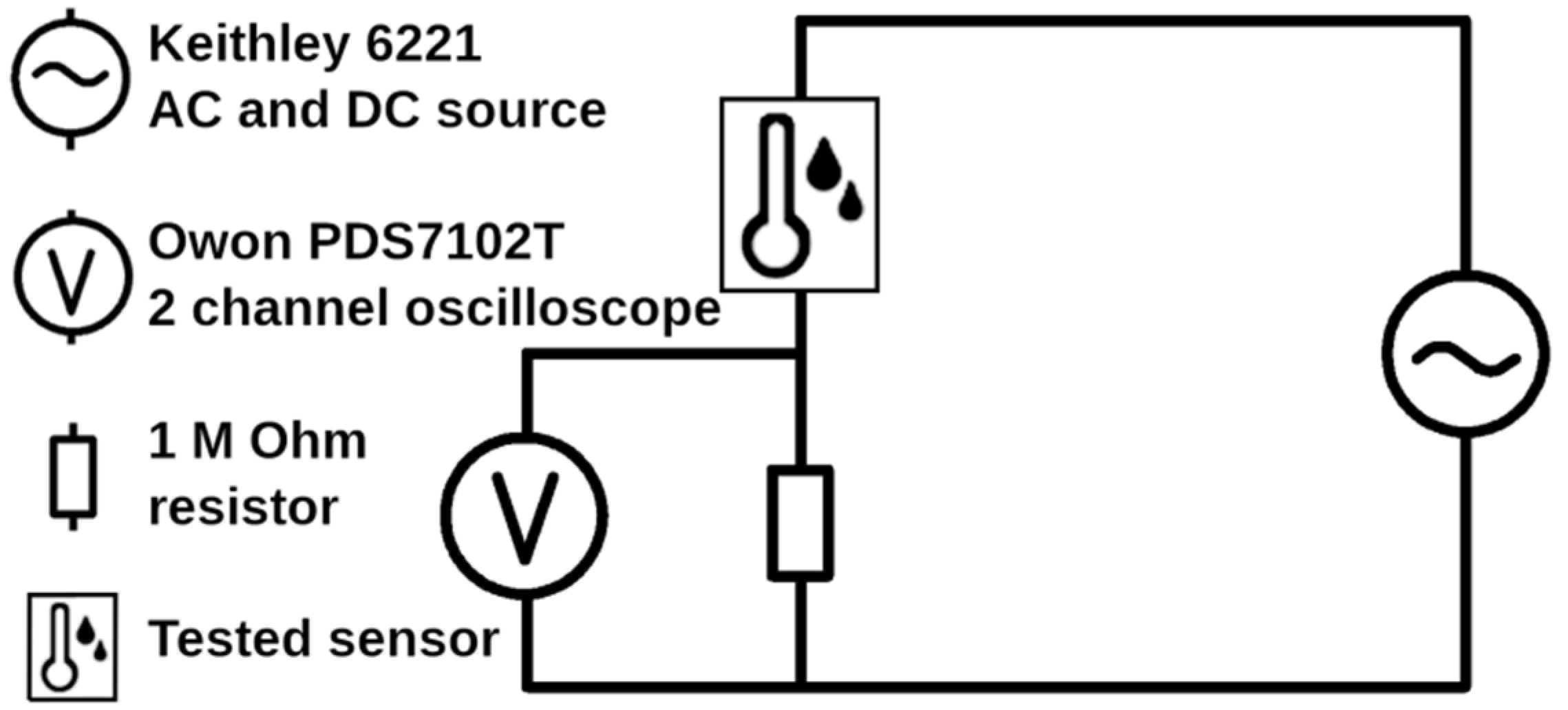
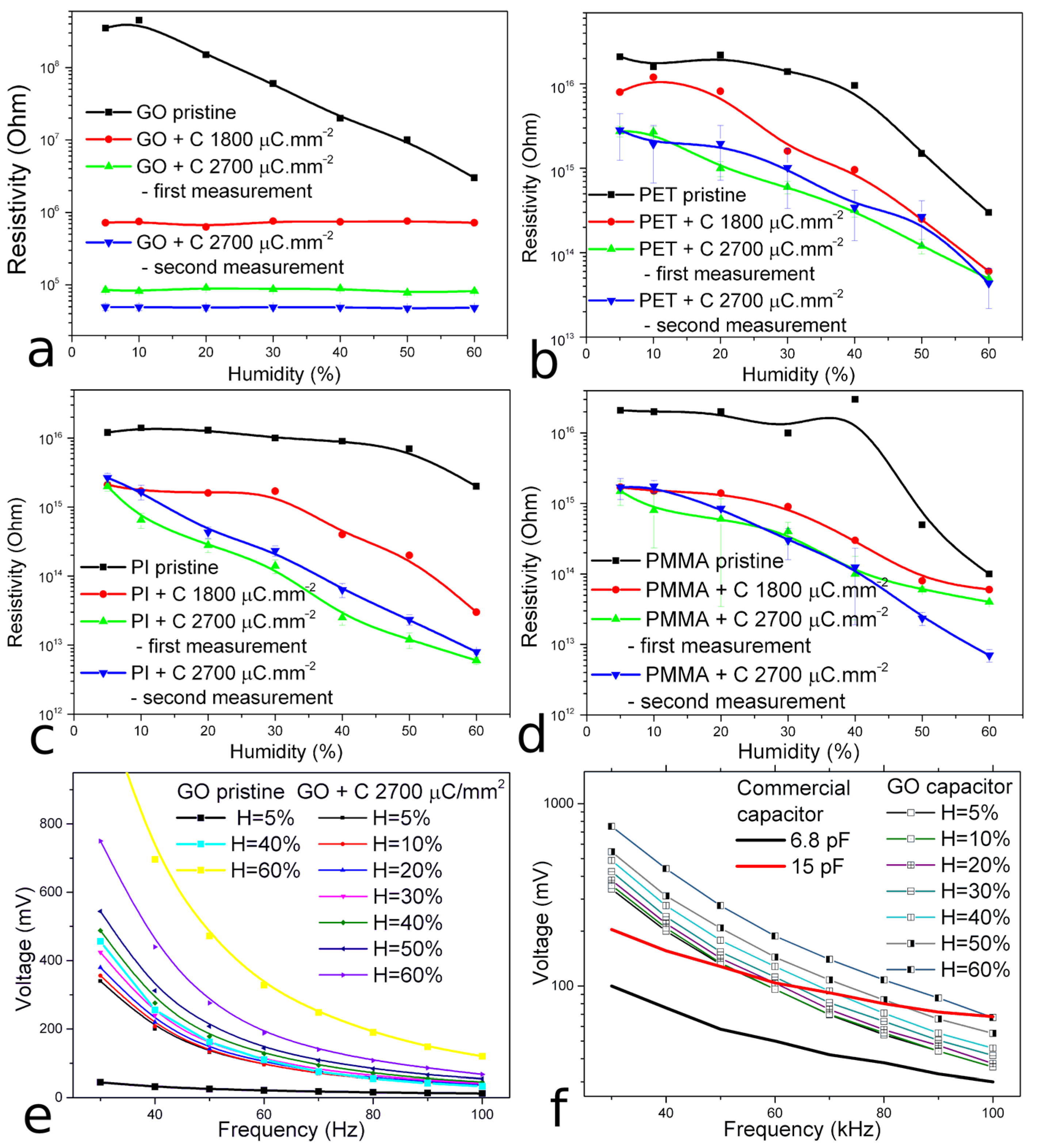
3.6. Sensing Properties of Prepared Microstructures
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Nie, J.; Wu, Y.; Huang, Q.; Joshi, N.; Li, M.; Meng, X.; Zheng, S.; Zhang, M.; Mi, B.; Lin, L. Dew Point Measurement Using a Carbon-Based Capacitive Sensor with Active Temperature Control. ACS Appl. Mater. Interfaces 2019, 11, 1699–1705. [Google Scholar] [CrossRef]
- Bi, H.; Yin, K.; Xie, X.; Ji, J.; Wan, S.; Sun, L.; Terrones, M.; Dresselhaus, M.S. Ultrahigh humidity sensitivity of graphene oxide. Sci. Rep. 2013, 3, 2714. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, L.; Yang, Y.; Huang, Q.; Li, D.; Zeng, D. A review on two-dimensional materials for chemiresistive- and FET-type gas sensors. Phys. Chem. Chem. Phys. 2021, 23, 15420–15439. [Google Scholar] [CrossRef]
- Pi, S.; Zhang, X.; Cui, H.; Chen, D.; Zhang, G.; Xiao, S.; Tang, J. Facile Fabrication of Au Nanoparticles/Tin Oxide/Reduced Graphene Oxide Ternary Nanocomposite and Its High-Performance SF6 Decomposition Components Sensing. Front. Chem. 2019, 7, 476. [Google Scholar] [CrossRef]
- Boudanen, J.; Stenmassl, M.; Endres, H.E.; Drost, A.; Eisele, I.; Kutter, C.; Mülle-Buschbaum, P. Polyimide-Based Capacitive Humidity Sensor. Sensors 2018, 18, 1516. [Google Scholar] [CrossRef]
- Wu, Y.; Huang, Q.; Nie, J.; Liang, J.; Joshi, N.; Hayasaka, T.; Zhao, S.; Zhang, M.; Wang, X.; Lin, L. All-Carbon Based Flexible Humidity Sensor. J. Nanosci. Nanotechno. 2019, 19, 5310–5316. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Y.; Yang, Z.; Hu, N. Review of recent progress on graphene-based composite gas sensors. Ceram. Int. 2021, 47, 16367–16384. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, A.; Han, Y.; Li, T. Sensors based on conductive polymers and their composites: A review. Polym. Int. 2020, 69, 7–17. [Google Scholar] [CrossRef]
- Zhi, C.; Chi, L. Humidity Sensors: A Review of Materials and Mechanisms. Sens. Lett. 2005, 3, 274–295. [Google Scholar]
- Borini, S.; White, R.; Wei, D.; Astley, M.; Haque, S.; Spigone, E.; Harris, N.; Kivioja, J.; Ryhänen, T. Ultrafast Graphene Oxide Humidity Sensors. ACS Nano 2013, 12, 11166–11173. [Google Scholar] [CrossRef]
- Basu, S.; Bhattacharyya, P. Recent developments on graphene and graphene oxide based solid state gas sensors. Sens. Actuator 2012, 173, 1–21. [Google Scholar] [CrossRef]
- Lee, H.; Lee, S.; Jung, S.; Lee, J. Nano-grass polyimide-based humidity sensors. Sens. Actuator 2009, 154, 2–8. [Google Scholar] [CrossRef]
- Packirisamy, M.; Stiharu, I.; Li, X.; Rinaldi, G. A polyimide based resistive humidity sensor. Sens. Rev. 2005, 25, 271–276. [Google Scholar] [CrossRef]
- Kuroiwa, T.; Hayashi, T.; Ito, A.; Matsuguchi, M.; Sadaoka, Y.; Sakai, Y. A thin film polyimide based capacitive type relative humidity sensor. Sens. Actuators B-Chem. 1993, 13, 59–91. [Google Scholar] [CrossRef]
- Harith, Z.; Zain, H.A.A.; Batumalay, M.; Harun, S.W. A study on relative humidity sensors using PVA and PMMA coating. J. Phys. Conf. Ser. 2019, 1371, 012027. [Google Scholar] [CrossRef]
- Liehr, S.; Breithaupt, M.; Krebber, K. Distributed Humidity Sensing in PMMA Optical Fibers at 500 nm and 650 nm Wavelengths. Sensors 2017, 17, 738. [Google Scholar] [CrossRef]
- Su, P.G.; Sun, Y.L.; Lin, C.C. Humidity sensor based on PMMA simultaneously doped with two different salts. Sens. Actuators B-Chem. 2006, 113, 883–886. [Google Scholar] [CrossRef]
- Zhang, D.; Tong, J.; Xia, B.; Xue, Q. Ultrahigh performance humidity sensor based on layer-by-layer self-assembly of graphene oxide/polyelectrolyte nanocomposite film. Sens. Actuators B-Chem. 2014, 203, 263–270. [Google Scholar] [CrossRef]
- Reddy, A.S.G.; Narakathu, B.B.; Atashbar, M.Z.; Rebros, M.; Rebrosova, E.; Joyce, M.K. Fully Printed Flexible Humidity Sensor. Procedia Eng. 2011, 25, 120–123. [Google Scholar] [CrossRef]
- Zhang, D.; Chang, H.; Li, P.; Liu, R.; Xue, Q. Fabrication and characterization of anultrasensitive humidity sensor based on metal oxide/graphene hybrid nano-composite. Sens. Actuators B-Chem. 2016, 225, 233–240. [Google Scholar] [CrossRef]
- Kwon, S.N.; Jung, C.H.; Na, S.I. Electron-beam-induced reduced graphene oxide as an alternative hole-transporting interfacial layer for high-performance and reliable polymer solar cells. Org. Electron. 2016, 34, 67–74. [Google Scholar] [CrossRef]
- Jung, J.M.; Jung, C.H.; Oh, M.S.; Hwang, I.T.; Jung, C.H.; Shin, K.; Hwang, J.; Park, S.H.; Choi, J.H. Rapid, facile, and eco-friendly reduction of graphene oxide by electron beam irradiation in an alcohol–water solution. Mater. Lett. 2014, 126, 151–153. [Google Scholar] [CrossRef]
- Resta, V.; Quarta, G.; Farella, I.; Maruccio, L.; Cola, A.; Calcagnile, L. Optical and electrical properties of polycarbonate layers implanted by high energy Cu ions. Nucl. Instrum. Meth. B 2014, 331, 168–171. [Google Scholar] [CrossRef]
- Popok, V. Ion implantatio of polymers: Formation of nanoparticulate materials. Rev. Adv. Mater. Sci. 2012, 30, 1–26. [Google Scholar]
- Döbeli, M.; Jones, T.J.; Lee, A.; Livi, R.P.; Tombrelo, T.A. Electrical conductivity of iron-irradiated carbon. Radiat. Eff. Defects Solids 1991, 118, 325–339. [Google Scholar] [CrossRef]
- Malinský, P.; Mackova, A.; Florianova, M.; Cutroneo, M.; Hnatowicz, V.; Bohacova, M.; Szokolova, K.; Bottger, R.; Sofer, Z. The Structural and Compositional Changes of Graphene Oxide Induced by Irradiation With 500 keV Helium and Gallium Ions. Phys. Stat. Sol. B 2019, 256, 1800409. [Google Scholar] [CrossRef]
- Malinský, P.; Cutroneo, M.; Mackova, A.; Hnatowicz, V.; Florianova, M.; Bohacova, M.; Bousa, D.; Sofer, Z. Graphene oxide layers modified by irradiation with 1.2 MeV He+ ions. Surf. Coat. Tech. 2018, 342, 220–225. [Google Scholar] [CrossRef]
- Cutroneo, M.; Havranek, V.; Mackova, A.; Malinsky, P.; Torrisi, L.; Lorincik, J.; Luxa, J.; Szokolova, K.; Sofer, Z.; Stammers, J. Localized deoxygenation of graphene oxide foil by ion microbeam writing. Vacuum 2019, 163, 10–14. [Google Scholar] [CrossRef]
- Jankovsky, O.; Simak, P.; Luxa, J.; Sedmidubsky, D.; Tomandl, I.; Mackova, A.; Miksova, R.; Malinsky, P.; Pumera, M.; Sofer, Z. Definitive insight into the graphite oxide reduction mechanism by deuterium labeling. ChemPlusChem 2015, 80, 1399–1407. [Google Scholar] [CrossRef]
- Available online: http://www.goodfellow.com/ (accessed on 22 September 2022).
- Martienssen, W.; Warlimont, H. Springer Handbook of Condensed Matter and Materials Data; Springer: Berlin/Heidelberg, Germany, 2005; ISBN 978-3-540-3043. [Google Scholar]
- Mayer, M. SIMNRA User’s Guide, Report IPP 9/113; Max-Planck-Institut fur Plasmaphysik: Garching, Germany, 1997. [Google Scholar]
- Malinsky, P.; Romanenko, A.; Havranek, V.; Stammers, J.H.; Hnatowicz, V.; Cutroneo, M.; Novak, J.; Slepicka, P.; Svorcik, V.; Szokolova, K.; et al. Microcapacitors on graphene oxide and synthetic polymers prepared by microbeam lithography. Appl. Surf. Sci. 2020, 528, 146802. [Google Scholar] [CrossRef]
- Thomaz, R.S.; Papaléo, R.M. Ion Beam Modification of Poly (methyl methacrylate). In Radiation Effects in Polymeric Materials; Springer Series on Polymer and Composite Materials; Kumar, V., Chaudhary, B., Sharma, V., Verma, K., Eds.; Springer: Cham, Switzerland, 2019; pp. 113–139. [Google Scholar]
- Liu, Y.; Chen, Q.; Du, X.; Li, X.; Li, P. Surface modification of polyethylene terephthalate films by direct fluorination. AIP Adv. 2018, 8, 125333. [Google Scholar] [CrossRef]
- Jamalzadeh, M.; Sobkowicz, M.J. Review of the effects of irradiation treatments on poly(ethylene terephthalate). Polym. Deg. Stab. 2022, 206, 110191. [Google Scholar] [CrossRef]
- Hwang, I.T.; Kuk, I.S.; Jung, C.H.; Choi, J.H.; Nho, Y.C.; Lee, Y.M. Efficient Immobilization and Patterning of Biomolecules on Poly(ethylene terephthalate) Films Functionalized by Ion Irradiation for Biosensor Applications. ACS Appl. Mater. Interfaces 2011, 3, 2235–2239. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Q.S.; Bashir, S.; Jalil, S.A.; Shabbir, M.K.; Akram, M.; Khalid, A.; Yaseen, N.; Arshad, A. Surface, electrical and mechanical modifications of PMMA after implantation with laser produced iron plasma ions. Nucl. Instrum. Meth. B 2016, 378, 1–7. [Google Scholar] [CrossRef]
- Popok, V.N.; Zarko, I.I.; Khaibullin, R.I.; Stepanov, A.L.; Hnatowicz, V.; Mackova, A.; Prasalovich, S.V. Radiation induced change of polyimide properties under high fluence and high ion current density implantation. Appl. Phys. A 2004, 78, 1067–1072. [Google Scholar] [CrossRef]
- Kochumalayil, J.J.; Meiser, A.; Soldera, F.; Possart, W. Focused ion beam irradiation—morphological and chemical evolution in PMMA. Surf. Interface Anal. 2009, 41, 412–420. [Google Scholar] [CrossRef]
- Puttaraksa, N.; Norarat, R.; Laitinen, M.; Sajavaara, T.; Sinkgkarat, S.; Whitlow, H.J. Litography exposure characteristics of poly(methyl methacrylate) (PMMA) for carbon, helium and hydrogen ions. Nucl. Instrum. Meth. B 2012, 272, 162–164. [Google Scholar] [CrossRef]
- Romanenko, O.; Havranek, V.; Malinsky, P.; Slepicka, P.; Stammers, J.; Svorcik, V.; Mackova, A.; Fajstavr, D. Effect of irradiation conditions by swift heavy ions on the microstructure and composition of PMMA. Nucl. Instrum. Meth. B 2019, 461, 175–180. [Google Scholar] [CrossRef]
- Constantini, J.M.; Couvreur, F.; Salvetat, J.P.; Bouffard, S. Micro-Raman study of the carbonization of polyimide induced by swift heavy ion irradiations. Nucl. Instrum. Meth. B 2002, 194, 132–140. [Google Scholar] [CrossRef]
- Viana, M.M.; Lima, M.C.F.S.; Forsythe, J.C.; Gangoli, V.S.; Cho, M.; Cheng, Y.; Silva, G.G.; Wong, M.S.; Caliman, V. Facile Graphene Oxide Preparation by Microwave-Assisted Acid Method. J. Braz. Chem. Soc. 2015, 26, 978–984. [Google Scholar] [CrossRef]
- Kavetskyy, T.; Nowak, J.; Borc, J.; Rusnak, J.; Šauša, O.; Stepanov, A.L. Carbonization in boron-ion implanted polymethylmethacrylate as revealed from Raman spectroscopy and electrical measurements. Spectrosc. Lett. 2015, 49, 5–10. [Google Scholar] [CrossRef]
- Lippert, T.H.; Zimmermann, F.; Wokaun, A. Surface Analysis of Excimer-Laser-Treated Polyethylene-Terephthalate by Surface-Enhanced Raman Scattering and X-Ray Photoelectron Spectroscopy. Appl. Spectrosc. 1993, 47, 1931–1942. [Google Scholar] [CrossRef]
- Hareesh, A.K.; Joshi, R.P.; Shateesh, B.; Kandasami, A.; Kanjilal, D.; Late, D.; Dahiwale, S.; Bhoraskar, V.; Haram, S.; Dhole, S.D. Reduction of graphene oxide by 100 MeV Au ion irradiation and its application as H2 O2 sensor. J. Phys. D Appl. Phys. 2015, 48, 365105. [Google Scholar] [CrossRef]
- Mathew, S.; Chan, T.K.; Gopinadhan, K.; Barman, A.R.; Breese, M.B.H.; Dhar, S.; Shen, R.S.; Venkatesan, T.; Thong, J.L.T. Mega-electron-volt proton irradiation on supported and suspended graphene: A Raman spectroscopic layer dependent study. J. Appl. Phys. 2011, 110, 084309. [Google Scholar] [CrossRef]
- Ambrosi, A.; Bonanni, A.; Sofer, Z.; Cross, J.S.; Pumera, M. Electrochemistry at chemically modified graphene. Chemistry 2011, 17, 10763–10770. [Google Scholar] [CrossRef]
- Babtista, D.L.; Zawislak, F.C. Hard and sp2-rich amorphous carbon structure formed by ion beam irradiation of fullerene, a-C and polymeric a-C: H films. Diam. Relat. Mater. 2004, 13, 1791–1801. [Google Scholar] [CrossRef]
- Arif, S.; Rafique, M.S.; Saleemi, F.; Sagheer, R.; Naab, F.; Toader, O.; Mahmood, A.; Rashid, R.; Mahmood, M. Influence of 400 keV carbon ion implantation on structural, optical and electrical properties of PMMA. Nucl. Instrum. Meth. B 2015, 358, 236–244. [Google Scholar] [CrossRef]
- Pei, S.; Cheng, H.M. The reduction of graphene oxide. Carbon 2012, 50, 3210–3228. [Google Scholar] [CrossRef]
- Malinsky, P.; Mackova, A.; Miksova, R.; Kovacikova, H.; Cutroneo, M.; Luxa, J.; Bousa, D.; Strochova, B.; Sofer, Z. Graphene oxide layers modified by light energetic ions. Phys. Chem. Chem. Phys. 2017, 19, 10282–10291. [Google Scholar] [CrossRef]
- Malinsky, P.; Cutroneo, M.; Mackova, A.; Hnatowicz, V.; Szökölová, K.; Bohačová, M.; Luxa, J.; Sofer, Z. Graphene oxide layers modified by irradiation with 1.0 MeV Au+ ions. Surf. Interface Anal. 2018, 50, 1110–1115. [Google Scholar] [CrossRef]
- Yuge, R.; Zhang, M.; Tomonari, M.; Yoshitake, T.; Iijima, S.; Yudasaka, M. Site Identification of Carboxyl Groups on Graphene Edges with Pt Derivatives. ACS Nano 2008, 2, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.W.; Yung, K.C.; Xie, C.S. XPS investigation of the chemical characteristics of Kapton films ablated by a pulsed TEA CO2 laser. Surf. Coat. Tech. 2002, 153, 210–216. [Google Scholar] [CrossRef]
- Khomiakova, N.; Hanuš, J.; Kuzminova, A.; Kylián, O. Investigation of Wettability, Drying and Water Condensation on Polyimide (Kapton) Films Treated by Atmospheric Pressure Air Dielectric Barrier Discharge. Coatings 2020, 10, 619. [Google Scholar] [CrossRef]
- Pletincx, S.; Marcoen, K.; Trotochaud, L.; Fockaer, L.L.; Mol, J.M.C.; Head, A.R.; Karslioglu, O.; Bluhm, H.; Terryn, H.; Haufman, T. Unravelling the Chemical Influence of Water on the PMMA/Aluminum Oxide Hybrid Interface In Situ. Nature 2017, 7, 13341. [Google Scholar] [CrossRef]
- Abdel-Fattah, E. Surface Activation of Poly(Methyl Methacrylate) with Atmospheric Pressure Ar+H2O Plasma. Coatings 2019, 9, 228. [Google Scholar] [CrossRef]
- Song, S.; Wan, C.; Zhang, Y. Non-covalent functionalization of graphene oxide by pyrene-block copolymers for enhancing physical properties of poly(methyl methacrylate). RSC Adv. 2015, 97, 79947–79955. [Google Scholar] [CrossRef]
- Ektessabi, A.M.; Hakamata, S. XPS study of ion beam modified polyimide films. Thin. Solid. Films. 2000, 377–378, 621–625. [Google Scholar] [CrossRef]
- Pistillo, B.R.; Menguelti, K.; Arl, D.; Leturcq, R.; Lenoble, D. PRAP-CVD: A Novel Technique to Deposit Intrinsically Conductive Polymers. In Polymerization; Cankaya, N., Ed.; Intechopen: London, UK, 2018; ISBN 978-953-51-3747-4. [Google Scholar]
- Gonzales, E.; Barankin, M.D.; Guschl, P.C.; Hicks, R.F. Remote Atmospheric-Pressure Plasma Activation of the Surfaces of Polyethylene Terephthalate and Polyethylene Naphthalate. Langmuir 2008, 24, 12636–12643. [Google Scholar] [CrossRef]
- Wu, Z.; He, J.; Yang, H.; Yang, S. Progress in Aromatic Polyimide Films for Electronic Applications: Preparation, Structure and Properties. Polymers 2022, 14, 1269. [Google Scholar] [CrossRef]
- Cheng, B.H.; Tian, B.X.; Xie, C.C.; Xiao, Y.H.; Lei, S.J. Highly sensitive humidity sensor based on amorphous Al2 O3 nanotubes. J. Mater. Chem. 2011, 21, 1907–1912. [Google Scholar] [CrossRef]
- Yoo, K.P.; Lim, L.T.; Mim, N.K.; Lee, M.J.; Lee, C.J.; Park, C.W. Novel resistive-type humidity sensor based on multiwall carbon nanotube/polyimide composite films. Sens. Actuators 2010, 145, 120–125. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C (at. %) | O (at. %) | H (at. %) | N (at. %) | C/O | C/H | |
|---|---|---|---|---|---|---|
| PET pristine | 57.7 ± 1.9 | 6.0 ± 0.4 | 36.3 ± 2.3 | 9.6 | 1.6 | |
| PET 3.750 × 1014 cm−2 | 67.4 ± 2.2 | 6.4 ± 0.4 | 26.2 ± 1.7 | 10.5 | 2.6 | |
| PET 5.625 × 1014 cm−2 | 65.2 ± 2.1 | 9.5 ± 0.5 | 25.4 ± 1.6 | 6.9 | 2.6 | |
| PI pristine | 56.4 ± 1.8 | 12.8 ± 0.7 | 25.6 ± 1.7 | 5.2 ± 0.3 | 4.4 | 2.2 |
| PI 3.750 × 1014 cm−2 | 61.7 ± 2.0 | 9.5 ± 0.6 | 23.6 ± 1.6 | 5.2 ± 0.3 | 6.5 | 2.6 |
| PI 5.625 × 1014 cm−2 | 62.4 ± 2.0 | 9.2 ± 0.5 | 23.0 ± 1.5 | 5.4 ± 0.3 | 6.8 | 2.7 |
| PMMA pristine | 54.2 ± 1.8 | 2.7 ± 0.3 | 43.0 ± 2.8 | 20.0 | 1.3 | |
| PMMA 3.750 × 1014 cm−2 | 62.6 ± 2.0 | 8.3 ± 0.8 | 29.0 ± 1.9 | 7.5 | 2.2 | |
| PMMA 5.625 × 1014 cm−2 | 74.2 ± 2.4 | 7.6 ± 0.7 | 18.0 ± 1.2 | 9.8 | 4.1 | |
| GO pristine | 68.0 ± 2.5 | 19.0 ± 1.0 | 9.0 ± 0.7 | 3.0 ± 0.2 | 3.6 | 7.6 |
| GO 3.750 × 1014 cm−2 | 70.0 ± 2.6 | 18.0 ± 0.9 | 8.0 ± 0.6 | 4.0 ± 0.4 | 3.9 | 8.8 |
| GO 5.625 × 1014 cm−2 | 71.0 ± 2.6 | 16.0 ± 0.8 | 8.0 ± 0.6 | 3.0 ± 0.2 | 4.4 | 8.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malinský, P.; Romanenko, O.; Havránek, V.; Cutroneo, M.; Novák, J.; Štěpanovská, E.; Mikšová, R.; Marvan, P.; Mazánek, V.; Sofer, Z.; et al. Graphene Oxide and Polymer Humidity Micro-Sensors Prepared by Carbon Beam Writing. Polymers 2023, 15, 1066. https://doi.org/10.3390/polym15051066
Malinský P, Romanenko O, Havránek V, Cutroneo M, Novák J, Štěpanovská E, Mikšová R, Marvan P, Mazánek V, Sofer Z, et al. Graphene Oxide and Polymer Humidity Micro-Sensors Prepared by Carbon Beam Writing. Polymers. 2023; 15(5):1066. https://doi.org/10.3390/polym15051066
Chicago/Turabian StyleMalinský, Petr, Oleksander Romanenko, Vladimír Havránek, Mariapompea Cutroneo, Josef Novák, Eva Štěpanovská, Romana Mikšová, Petr Marvan, Vlastimil Mazánek, Zdeněk Sofer, and et al. 2023. "Graphene Oxide and Polymer Humidity Micro-Sensors Prepared by Carbon Beam Writing" Polymers 15, no. 5: 1066. https://doi.org/10.3390/polym15051066
APA StyleMalinský, P., Romanenko, O., Havránek, V., Cutroneo, M., Novák, J., Štěpanovská, E., Mikšová, R., Marvan, P., Mazánek, V., Sofer, Z., & Macková, A. (2023). Graphene Oxide and Polymer Humidity Micro-Sensors Prepared by Carbon Beam Writing. Polymers, 15(5), 1066. https://doi.org/10.3390/polym15051066